











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Las epilepsias mioclónicas progresivas (EMP) son un grupo de enfermedades neurodegenerativas raras que engloban una de las formas más discapacitantes de epilepsias. Tienen heterogeneidad clínica y genética, pero comparten como característica común la presencia de mioclonías de acción, crisis epilépticas y deterioro neurológico progresivo. Este deterioro neurológico puede expresarse como deterioro cognitivo en la mayoría de casos, pero también como ataxia o neuropatía 1,2,3.
La edad de presentación más común es en la infancia tardía o la adolescencia, pero puede afectar a personas de todas las edades 1. El cuadro clínico inicial de estas enfermedades puede ser confundido con otras condiciones clínicas más frecuentes, dificultando su diagnóstico, en especial con otro grupo de epilepsias generalizadas de origen genético, como la epilepsia mioclónica juvenil 1,3.
Las mioclonías que dan nombre a estas enfermedades son contracciones súbitas y muy breves de los músculos que suceden por descargas anómalas en el sistema nervioso central. El origen de estas mioclonías suele ser cortical, pudiendo ser en algunos casos cortico-subcortical. Se distinguen por ser desencadenadas por actividad muscular voluntaria o por algunos estímulos sensitivos o emociones 1,2,3. En cuanto a las crisis epilépticas, predominan las tónico-clónicas generalizadas, pero puede haber otros tipos como de ausencia, tónicas y focales. Suele haber enlentecimiento de fondo en el electroencefalograma(EEG). Lamentablemente, no suelen responder al tratamiento antiepiléptico 1.
Actualmente se conoce la etiología genética de la mayoría de las enfermedades agrupadas dentro de las EMP. La herencia suele ser autosómica recesiva, aunque hay enfermedades que tienen herencia autosómica dominante (como la atrofia dentato-rubro- pálido-luisiana) o mitocondrial (como la epilepsia mioclónica con fibras rojas rasgadas o MERRF). Por esto es importante identificar genéticamente el síndrome, ya que algunos se benefician de tratamientos específicos como la enfermedad de Gaucher o de Tay- Sachs. Sin embargo, aún hay casos de pacientes en los que no se identifican variantes genéticas patogénicas o probablemente patogénicas que permitan clasificarlos en alguna de estas enfermedades 1,2,3. El pronóstico de estos pacientes es variable según cada enfermedad, pero suele ser malo, llevando a los pacientes que la sufren a la discapacidad o a una muerte temprana 1. En el presente artículo se hará una revisión de las formas de epilepsia mioclónica progresiva más frecuentes. En la Tabla 1 se puede apreciar un resumen de algunas características distintivas de cada enfermedad discutida a continuación.
Tabla 1 Características de epilepsias mioclónicas progresivas
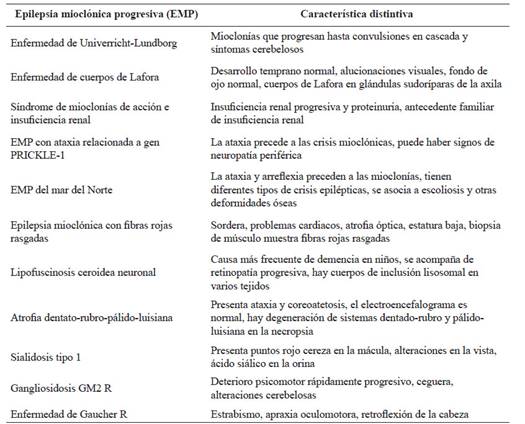
Fuente: elaboración a partir de información en Malek N, Stewart W, Greene J. The progressive myoclonic epilepsies. Pract Neurol. 2015;15(3):164-171.
Enfermedad de Unverricht-Lundborg (EMP1)
También llamada epilepsia mioclónica progresiva tipo 1 (EMP1).
Genética: Es una enfermedad hereditaria autosómica recesiva. Es causada por variantes en el gen CSTB, que lleva a una función deficiente de cistatina B, de la superfamilia de inhibidores de proteasas. La cistatina protege a las células de proteasas endógenas, como las catepsinas, y su deficiencia causa hiperexcitabilidad y alteración de la función neuronal de redes corticales. Se conocen 14 mutaciones diferentes que lo pueden causar 1,2,3,4.
Epidemiología: Es la EMP más frecuente a nivel mundial. La mayor prevalencia de EMP1 se describe en Finlandia con uno en 20 000 nacimientos. No hay datos de su prevalencia en otros países, pero se está diagnosticando más seguido en epilepsias resistentes al tratamiento médico en la adolescencia. Inicia a los 6-16 años, con mayor incidencia a los 12-13 años 1,2,5.
Clínica: Se caracteriza por mioclonías sensitivas a estímulo y crisis tónico-clónicas generalizadas (CTCG). El inicio de la enfermedad suele ser con CTCG, predominantemente al despertar o durante el sueño. Además, presenta mioclonías que también predominan al despertar 1,6. Con la progresión de la enfermedad, las mioclonías progresan, se hacen relacionadas al movimiento y aumentan con el estrés. Característico de esta enfermedad es que pueden desencadenarse por estímulos sensoriales como las luces o el tacto, o por el sentimiento de sorpresa. También pueden llevar a convulsiones en cascada, episodios que empiezan con mioclonías que aumentan de intensidad y violencia hasta llegar a una CTCG breve. No aparecen durante el sueño o el reposo. Pueden llevar al paciente a la discapacidad, postrándolo en una silla de ruedas 1,2,6. Además, el paciente desarrolla síntomas cerebelosos como ataxia, incoordinación, tremor de intención y disartria. Las alteraciones cognitivas suelen ser ausentes o ser leves a moderadas, pueden mejorar levemente con los años. Es común que aparezca depresión, con conductas suicidas 1,6.
Diagnóstico: Estudio genético que identifique variantes patogénicas o probablemente patogénicas en el gen EMP1. Solo se debe hacer en un paciente con sospecha clínica por historia y examen físico, con hallazgos en el EEG que lo diferencien de otros tipos de EMP, o como parte de la reevaluación de un caso de epilepsia mioclónica juvenil que no responde al tratamiento. Si esta prueba es positiva, se confirma el diagnóstico. Si es negativa pero la sospecha clínica, tras una discusión con un neurólogo con conocimiento del tema, es muy alta, se puede hacer el diagnóstico, a pesar de la prueba negativa 6. En el EEG hay descargas epileptiformes generalizadas en la mayoría de pacientes, la mitad muestra potenciales evocados somatosensoriales gigantes 1.
Manejo: Recordar que la enfermedad varía de paciente a paciente y que su progresión es limitada. Para el tratamiento de las epilepsias, se usa típicamente valproato. El piracetam es un agente que puede aliviar las mioclonías. Se recomienda no usar lamotrigina ya que puede agravar las mioclonías. Tampoco se debe usar fenitoína, que empeora las mioclonías y precipita aparición de signos cerebelosos y atrofia cerebelar. Otros antiepilépticos que se pueden usar son fenobarbital o primidona (que causan deterioro cognitivo), levetiracetam, topiramato y zonisamida. No hay evidencia que haya beneficio con el uso de carbamazepina, oxcarbazepina, eslicarbazepina, gabapentina, pregabalina, vigabatrin o lacosamida. Se pueden usar benzodiazepinas para calmar al paciente, con cuidado para evitar la tolerancia. Se pueden usar regímenes de hasta 3 a 5 antiepilépticos diferentes. En el caso de mioclonías intratables, se puede probar con estimulación del nervio vago o estimulación cerebral profunda, ambas con evidencia limitada. No hay tratamiento genético específico para esta enfermedad 3,5,6.
Pronóstico: La evolución suele ser una progresión de la enfermedad por 5-10 años, tras lo cual se estabiliza. El resultado en la persona tiene un gran rango: desde un adulto independiente con vida activa a una persona dependiente de silla de ruedas o postrados en cama. La muerte puede ser por suicidios o muerte súbita e inesperada en la epilepsia (SUDEP, por sus siglas en inglés) 6.
Enfermedad de cuerpos de Lafora
También llamada epilepsia mioclónica progresiva tipo II. Es un desorden del metabolismo de carbohidratos 1.
Genética: Es una enfermedad hereditaria autosómica recesiva. Causada por variantes en el gen EPM2A o en el gen EPM2B. El primero codifica la laforina, fosfatasa de especificidad dual implicada en la desfosforilación del glucógeno, y el segundo codifica la malina, una E3 ubiquitina ligasa implicada en la poliubiquitinación de proteínas relacionadas con el metabolismo del glucógeno. Se propone como un tercer gen el PRMD8, asociado a una enfermedad de inicio más temprano y que codifica una proteína responsable por la translocación de laforina y malina en el núcleo, su mutación causa una deficiencia de laforina y malina 1,2,7,8. La malina y laforina interactúan con la vía celular que protege contra la acumulación citoplasmática de poliglucosano. Su mutación lleva a la formación de cuerpos de inclusión poligucosados en tejidos neuronales, muscular, corazón, hígado, retina y otros; llamados cuerpo de Lafora, patognomónicos de esta enfermedad 1,2,7.
Epidemiología: La mayoría de pacientes diagnosticados tienen ascendencia del Mediterráneo, norte de África o Asia central. La prevalencia es cercana a 4 casos por 1 000 000 de personas. Suele iniciar a los 12-15 años, pero una variante más temprana inicia a los 5 años 1,7.
Clínica: Los primeros síntomas suelen ser crisis epilépticas occipitales focales o generalizadas, que pueden ser desencadenadas por la luz y se asocia a ceguera transitoria o alucinaciones visuales, pero el fondo de ojo es normal. También hay caídas por crisis de ausencia o de atonía y mioclonías al reposo o en respuesta a estímulos. De forma progresiva, aparecen y aumentan en frecuencia CTCG, y la mioclonía se vuelve casi continua, en especial en extremidades superiores e inferiores, postrando al paciente en silla de ruedas 1,8. El desarrollo psicomotor en los primeros años es normal. El deterioro neurológico y cognitivo es progresivo y rápido. Inicia con una disminución del rendimiento académico y progresa a afasia, apraxia, ataxia cerebelosa, disminución del tono muscular en general, espasticidad y ceguera cortical. Hay alteraciones cognitivas a nivel de atención, funciones ejecutivas, lenguaje, memoria y agnosia visual 1,8. También pueden aparecer síntomas psiquiátricos como alteraciones de conducta, depresión, apatía, confusión o psicosis 8.
Diagnóstico: Es basado en hallazgos clínicos, en el EEG e histológicos por la presencia de cuerpos de Lafora en una biopsia de glándulas sudoríparas de la axila. También se puede hacer un análisis genético que identifique variantes patogénicas o probablemente patognéticas en los genes EPM2A o EPM2B1,2,7,8. El EEG muestra grados variables de enlentecimiento de la actividad de fondo en la mayoría de pacientes, descargas epileptiformes en el 85% de pacientes, descargas focales en un tercio de pacientes, fotosensibilidad a estímulos de rápida frecuencia en un cuarto de pacientes, y potenciales evocados somatosensoriales gigantes en dos tercios de los pacientes 1.
Manejo: Los fármacos antiepilépticos más usados son el valproato, fenobarbital, benzodiazepinas, piracetam, levetiracetam y zonisamida. No se deben usar vigabatrina, carbamazepina, fenitoína, gabapentina, pregabalina, tiagabina y lamotrigina debido a que pueden agravar los síntomas de la enfermedad, Se puede usar perampanel, antagonista selectivo de receptores de glutamato tipo AMPA, mejora crisis epilépticas, mioclonías, disfunción cognitiva y neurológica. Otra opción es la estimulación de nervio vago. En animales, se ha visto beneficio de usar ácido 4-fenilbutírico, metformina y selenato sódico. Los primeros disminuyen el número y duración de crisis y el último mejora degeneración neuronal. Actualmente no hay tratamiento genético aprobado para esta enfermedad 3,8.
Pronóstico: Los pacientes terminan postrados en cama. Aproximadamente a los 10 años del inicio de síntomas se produce la muerte, por status epiléptico y neumonía aspirativa o complicaciones por la degeneración del sistema nervioso3 , 8.
Síndrome de mioclonías de acción - insuficiencia renal
El síndrome de falla renal con mioclonías, también llamada epilepsia mioclónica progresiva tipo 4 (EMP4). Es una forma distintiva de epilepsia mioclónica progresiva que se asocia a falla renal 1,2.
Genética: Es una enfermedad autosómica recesiva asociada a pérdida de función por variantes en el gen SCARB2, que codifica la proteína LIMP2. Esta es una glicoproteína localizada en membranas lisosomales. Su sobreexpresión por la mutación causa agrandamiento de endosomas y lisosomas, alterando la endocitosis 1,2,4,9.
Epidemiología: Típicamente inicia entre el final de la segunda y tercera década de vida (generalmente entre los 15-25 años), hay una forma variable de inicio tardío en la quinta o sexta década de vida, sin falla renal. Actualmente se conocen 38 personas afectadas, pertenecientes a 26 familias diferentes. Los primeros casos descritos ocurrieron en franceses canadienses, pero se sabe que su distribución geográfica es más amplia (1,2,3,9, 10).
Clínica: Existe heterogeneidad de genotipo- fenotipo. Los pacientes pueden presentarse primero con sintomatología renal o neurológica. Del lado renal destaca proteinuria y daño renal progresivo, hasta enfermedad renal crónica terminal. Se describió originalmente como glomeruloesclerosis focal, también puede ser una glomerulopatía colapsante. Del lado neurológico, suelen iniciar con tremores en los dedos y manos que se exacerban por realizar funciones motores finas, como escribir, y progresan a mioclonías relacionadas a la acción y mioclonías involuntarias al reposo, llevando finalmente a crisis tónico clónica generalizadas y epilepsia mioclónica que empeora con el tiempo. Las mioclonías son multifocales y suelen afectar tronco, extremidades y cara. Otros síntomas neurológicos incluyen ataxia, disartria por disfunción cerebelosa, alteraciones auditivas y neuropatía periférica desmielinizante. A diferencia de otras EMP, la función cognitiva no es afectada 1,2,3,4,9.
Diagnóstico: Se debe sospechar en pacientes con diagnóstico de epilepsia mioclónica que desarrolla falla renal. Estudio genético que identifique variantes patogénicas o probablemente patogénicas en el gen SCARB21,8. El electroencefalograma muestra anormalidades epileptiformes generalizadas y lenta progresión de actividad de fondo con el paso de los años. La estimulación de luz puede desencadenar ráfagas de ondas punta polipunta generalizadas, asociadas a mioclonías 4.
Manejo: Para el manejo de las crisis mioclónicas se usa un régimen múltiple que puede incluir valproato, clonazepam, barbitúricos y piracetam, con efectividad parcial y mejora de calidad de vida. Debe evitarse el uso de lamotrigina, carbamazepina y oxcarbazepina, ya que puede agravar las mioclonías de los pacientes. No existen terapias específicas para la enfermedad 3,9,10.
Pronóstico: La muerte sucede 7-15 años después del inicio de la enfermedad 4.
Epilepsia mioclónica progresiva con ataxia relacionada a gen PRICKLE-1
También llamada epilepsia mioclónica progresiva tipo 5 1.
Genética: Causada por variantes del gen de proteína de polaridad de la célula planar 1 (PRICKLE-1, por sus siglas en inglés). Las proteínas PRICKLE son constituyentes centrales de la vía de señalización celular planar que establece la polaridad celular durante el desarrollo embriológico1.
Epidemiología: La edad de inicio es 5-10 años. No se conoce su prevalencia, hay 3 grandes familias descendientes del medio oriente afectadas y algunas personas no relacionadas 1,11.
Clínica: Se caracteriza por crisis epilépticas mioclónicas, CTCG (relacionadas al sueño), ataxia y una función cognitiva normal. La mioclonía de acción afecta los músculos de las extremidades o bulbares, algunas veces con mioclonías espontáneas de músculos faciales causando una disartria marcada 1,11. La ataxia surge antes de las crisis mioclónicas, entre los 4 y 5 años. Las mioclonías de acción afectan los músculos de los miembros o bulbares, algunas veces con mioclonías espontáneas de músculos faciales causando una disartria marcada. Las crisis epilépticas pueden ser mioclónicas o CTCG, a predominio nocturno. Puede haber signos de neuropatía periférica, como hiporreflexia o signo de Babinski. No hay deterioro cognitivo 1,11.
Diagnóstico: En niños o adolescentes con crisis mioclónicas, CTCG, deterioro neurológico y cognición normal, primero se debe buscar la enfermedad de Univerricht-Lundborg y cuerpos de Lafora, ya que son mucho más comunes. Si la sospecha es alta, se puede realizar un estudio genético para gen PRICKLE1, que confirma el diagnóstico 11.
Manejo: El tratamiento antiepiléptico puede incluir las siguientes drogas: valproato, clonazepam, zonisamida, piracetam y leveritacetam. Se recomienda evitar dar fenitoína, carbamazepina y oxcarbazepina. No hay tratamientos específicos para esta enfermedad 3,11.
Pronóstico: Variable, ya que el paciente puede morir por complicaciones de un deterioro neurológico rápido o sobrevivir 9.
Epilepsia mioclónica progresiva del mar del Norte
También llamada epilepsia mioclónica progresiva tipo 6 (EMP6) o Ataxia mioclónica progresiva asociada al gen GOSR21.
Genética: Es causada por una variante en el gen del complejo receptor de Golgi SNAP 2 (GOSR2, por sus siglas en inglés). Este gen codifica una proteína de tráfico de membrana que transporta proteínas hacia los compartimentos del aparato de Golgi 1,3.
Epidemiología: Inicia a los 2 años. Llamada del Mar del Norte por ser la localización de los primeros pacientes descritos. Hasta el momento, solo se han descrito 20 casos de esta enfermedad. 1,2,12.
Clínica: La ataxia se inicia alrededor de los 2 años, junto con arreflexia, seguida de mioclonías a los 6-7 años y crisis epilépticas de diferentes tipos a los 13- 14 años. A la segunda década de vida han perdido la independencia ambulatoria por las crisis epilépticas. Los pacientes tienen múltiples tipos de crisis epilépticas, incluyendo CTCG, crisis de ausencia y atonías. Las mioclonías son generalizadas, altamente foto sensitivas, empeoran con la actividad o el estrés emocional, pero mínimas al reposo. Existe deterioro cognitivo en las etapas tardías de la enfermedad. La mayoría de los pacientes desarrollan escoliosis en la adolescencia, y a veces puede haber deformidades óseas, incluyendo pie cavo y sindactilia. Se puede encontrar elevación de creatina quinasa (media 700 UI) 1,13,14.
Diagnóstico: Identificación de la variante c.430G>T en el gen GOSR2 1,11.
Manejo: El manejo de la epilepsia se hace con valproato, zonisamida, piracetam y levetiracetam, no suele ser efectivo. No hay tratamientos específicos para esta enfermedad 3,13.
Pronóstico: Pacientes se vuelven dependientes de silla de ruedas o postrados en cama con la progresión de las crisis epilépticas y mueren jóvenes por complicaciones relacionadas a las mioclonías no controladas, comúnmente antes de los 30 años 13.
Epilepsia mioclónica con fibras rojas rasgadas (MERRF)
Llamado MERRF, por sus siglas en inglés. También es conocida como la Enfermedad mitocondrial multisistémica, por la apariencia característica de la biopsia de músculo 1.
Genética: Esta enfermedad es causada por mutaciones en el ADN mitocondrial. Se sabe que las mutaciones puntuales que alteran la codificación de ácido ribononucleico de lisina tRNALys3, gen MT-TK, es responsable del 80% de casos conocidos. El gen MT- TK es un gen transcriptor de ARN que tiene un papel crítico en la formación de proteínas involucradas en la fosforilación oxidativa. Existen variantes en los genes MT-TL1, MT-TH y MT-TS1 asociadas a este fenotipo, pero son menos frecuentes. Estas mutaciones llevan finalmente a una alteración de la función mitocondrial en las células 1,3.
Epidemiología: Su prevalencia se ha medido en países del norte de Europa: en la población adulta del norte de Finlandia es de 0-1,5 por 100 000 personas, y en la población adulta del norte de Inglaterra es de 0,39 por 100 000 personas 1,15.
Clínica: El inicio de la enfermedad se suele dar en la niñez. Los niños suelen tener un desarrollo normal hasta el inicio de los síntomas. El primer síntoma son las mioclonías. Otras manifestaciones comunes son la epilepsia generalizada, ataxia, debilidad muscular, demencia, estatura baja, pérdida de audición y atrofia ocular. Síntomas más raros son la cardiomiopatía con síndrome de Wolff-Parkinson-White, retinopatía pigmentaria, signos piramidales, oftalmoparesia, presencia de múltiples lipomas a predominio de cuello y tronco superior 1,3,4,5. En esta mutación es más común ver la presencia de mioclonías y ataxia que la epilepsia 2,5.
Diagnóstico: La sospecha se basa en cuatro síntomas canónicos: mioclonías, epilepsia generalizada, atrofia cerebelosa y fibras rojas rasgadas en una biopsia muscular. Esta se realiza usando la coloración tricrómica de Gomori. Un estudio genético que identifique variantes patogénicas o probablemente patogénicas en el gen MT-TK y otros genes mitocondriales relacionados. Se debe estudiar a pacientes que tienen síndrome de Ramsay-Hunt para descartar MERRF 1,4,15.
En el electroencefalograma de dos tercios de pacientes hay enlentecimiento de la actividad de fondo, con o sin descargas epileptiformes generalizadas, una minoría muestra potenciales evocados somatosensoriales gigantes. Puede haber neuropatía o miopatía coexistente 1.
Manejo: Las crisis epilépticas pueden manejarse con anticonvulsivantes como levetiracetam, clonazepam o zonisamida. Las mejores combinaciones son levetiracetam más clonazepam, topiramato, zonisamida o piracetam. No debe usarse valproato por causar deficiencia de carnitina y causar daño hepático, además de ser tóxico en enfermedades mitocondriales 1,15,16.
Pronóstico: Es variable, depende de cada paciente 15.
Lipofuscinosis ceroidea neuronal
Son un grupo heterogéneo de enfermedades neurodegenerativas, hereditarias asociadas a almacenamiento lisosomal 1.
Genética: Se conocen más de 400 variantes en 14 loci diferentes (CLN1 al 14) en humanos. Suelen tener herencia autosómica recesiva, aunque se han descrito formas autosómico dominantes en los fenotipos de aparición tardía. La lista de loci y genes identificados por fenotipo se resumen en la Tabla 2 1,2,4.
Tabla 2 locus de lipofuscinosis ceroidea neuronal y relación con fenotipo.
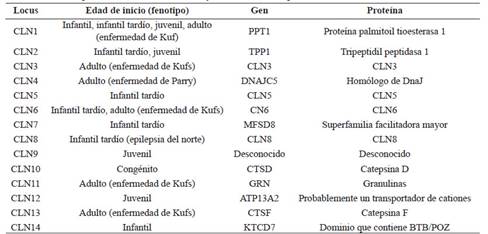
Basado en Malek N, Stewart W, Greene J. The progressive myoclonic epilepsies. Pract Neurol. 2015; 15(3): 164-171.
Epidemiología: Es la causa más frecuente de demencia en niños y el desorden lisosomal más común. Suele aparecer en sus diferentes formas entre el nacimiento y la adultez. El subtipo más raro es el que afecta a adultos, denominado enfermedad de Kufs. Su incidencia en Estados Unidos se estima en 1,6-2,4 por cada 100 000 habitantes 1,4,17.
Clínica: Se caracteriza por deterioro cognitivo y motor progresivo, crisis epilépticas tónico clónicas y mioclónicas progresivas, y muerte temprana. La mayoría tiene retinopatía que evoluciona a pérdida de visión y atrofia cerebelosa. Estos síntomas varían en edad y forma de inicio 1,2,4,17. Los fenotipos clínicos (previos a la era genética) se dividían en infantil, infantil tardío, juvenil y adulto, basados en la edad de inicio. Una nueva clasificación toma en cuenta los genes involucrados y la edad de inicio 1,2.
Diagnóstico: El análisis genético para la lipofuscinosis ceroidea neuronal está indicado en pacientes jóvenes con mioclonías, epilepsia y disminución de audición. Entre los hallazgos en la biopsia se encuentra lipopigmentación autofluorescente anormal de cuerpos de inclusión lisosomal depositados en cerebro, plexo mientérico, músculo y piel. Los depósitos lisosomales vistos en las biopsias varían según el tipo de variante de la enfermedad. También apunta al diagnóstico el hallazgo de un grado variable de atrofia cerebral en la resonancia magnética con cada variable teniendo una región preferente de atrofia 17. El electroencefalograma muestra grados variables de actividad de fondo enlentecida difusamente en el 95% de casos, descargas epileptiformes generalizadas en el 80% (Figura 1) 16. El 25% de pacientes tienen potenciales evocados somatosensoriales gigantes. Hay neuropatía axonal en el 30% de pacientes 1.
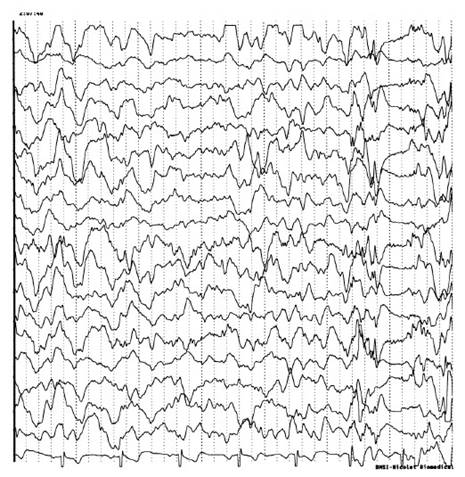
*Extraído de: Burneo JG, Arnold T, Palmer CA, Kuzniecky RI, Oh SJ, Faught E. Adult-onset neuronal ceroid lipofuscinosis (Kufs disease) with autosomal dominant inheritance in Alabama. Epilepsia. 2003; 44(6): 841- 846. Reproducido con el permiso de los autores.
Figura 1 electroencefalograma en el que se muestran mioclonías y actividad polipunta simultánea en un paciente con enfermedad de Kufs*
Manejo: Los antiepilépticos de elección son valproato y lamotrigina. No debe usarse lamotrigina por exacerbar crisis en CLN2. También se puede usar topiramato y levetiracetam. Usar benzodiazepinas con cuidado por sialorrea. Delusiones y alucinaciones se pueden tratar con neurolépticos atípicos como risperidona u olanzapina 17.
Pronóstico: Los afectados por CLN1 suelen fallecer alrededor de los 10 años de edad. Los afectados por CLN2 fallecen en la adolescencia por neumonía aspirativa. En CLN3 fallecen en segunda o tercera década de vida, en CLN5 sucede entre los 14 y 36 años, en CLN6 entre 5 y 12 años, los afectados por CLN8 fallecen entre 50 y 60 años 2,17.
Atrofia Dentato-rubro-pálido-luisiana
Es una ataxia espinocerebelosa tambien conocida como DRPLA, es una enfermedad caracterizada por epilepsia mioclónica progresiva, ataxia cerebelosa, coreoatetosis, mioclonías, demencia y síntomas psiquiátricos en varias combinaciones 1.
Genética: La enfermedad tiene una herencia autosómica dominante. Causado por una expansión inestable de repeticiones CAG en el exón 5 del gen ATN1en el cromosoma 12, que codifica tractos de poliglutamina. Es una enfermedad por expansión de repeticiones. Muestra el fenómeno de anticipación con formas más tempranas y severas en cada generación, la transmisión paterna resulta en una anticipación más prominente que la transmisión materna. La proteína DRPLA, o atrofina-1, es un coregulador de la transcripción 1.
Epidemiología: Es más común en población de ascendencia japonesa, con incidencia de 2-7 por 1 000 000 de habitantes. En estudios a nivel de América Latina, tiene frecuencia de 0,14-3,1% en cohortes en Brasil y Venezuela 19.
Clínica: Depende de la edad de inicio. Los pacientes con un fenotipo de epilepsia mioclónica progresiva tienen más expansiones (62-79 repeticiones) y menor edad de inicio (<20 años). Se caracterizan por iniciar con mioclonías, epilepsia, deterioro cognitivo, ataxia y coreoatetosis. El electroencefalograma (EEG) suele ser normal, algunas veces con descargas onda-punta atípicas y fotosensibilidad. Posterior a la muerte, los cambios neuropatológicos consisten en degeneración de sistemas dentado-rubro y pálido-luisiana 1,2. Los pacientes que se presentan sin epilepsia mioclónica progresiva tienen mayor edad de inicio (>20 años) y menor cantidad de repeticiones (54-67). Se caracteriza por ataxia, deterioro cognitivo y coreoatetosis 1,19. Diagnóstico: Es difícil por la heterogeneidad de la presentación, se puede confundir con otras ataxias espinocerebelosas. Se debe realizar pruebas genéticas en pacientes con un patrón de herencia autosómica dominante e historia familiar de deterioro cognitivo, demencia o alteraciones del movimiento. El estudio genético se basa en análisis de repeticiones CAG en el gen ATN119.
Manejo: El tratamiento que se da es de soporte multidisciplinario. No hay ensayos clínicos para evaluar la eficacia de antiepilépticos, pero se puede usar valproato, perampanel y zonisamida para el tratamiento de las crisis generalizadas. Para los movimientos coreoatetoicos y distónicos se usa tetrabenazina, risperidona, bromazepam y gabapentina. Para la ataxia se recomienda riluzol o amantadina 19.
Pronóstico: La esperanza de vida posterior al inicio de los síntomas es de 8 a 16 años 19.
Sialidosis tipo 1 (síndrome de mioclonías con punto rojo cereza o ST-1) y sialidosis tipo 2 (ST-2)
También llamada mucolipidosis. 1.
Genética: Es de herencia autosómica recesiva. La ST-1 y ST-2 son causadas por variantes del gen NEU1 en el cromosoma 6, que codifica la enzima α-N- acetilneuroaminidasa-1 (sialidasa). Estas variantes causa una deficiencia de esta enzima y acumulación de oligosacáridos sialidados en tejidos y excreción urinaria 1,20.
Epidemiología: Suele iniciar en la segunda o tercera década de vida. Su prevalencia se estima entre 1 cada 5 000 000 y 1 cada 15 000 000 de nacidos vivos 1.
Clínica: Tiene 2 formas clínicas, el tipo 1 es más leve y el tipo 2 más severa y de inicio más temprano, llamada sialidosis infantil. Se caracteriza por epilepsia mioclónica, alteraciones visuales, hiperreflexia, ataxia. Las mioclonías son el síntoma principal y se pueden precipitar por movimientos voluntarios, el pensamiento del movimiento, movimientos pasivos de articulaciones, luz, tacto o sonido. Siempre presentan puntos rojo cereza maculares, pero no son patognomónicos ya que también se ven en la oclusión de la arteria retinal central y enfermedades de almacenamiento metabólico como Tay-Sachs, Sandhoff, Niemann-Pick, Fabry y Gaucher, algunos de los cuales también tienen un fenotipo de epilepsia mioclónica 1,2,20.
La sialidosis tipo 2 puede presentar rasgos dismórficos (rasgos faciales toscos, tronco corto, tórax en tonel, deformidad vertebral y displasia esquelética o disostosis multiplex), retardo mental severo y a veces se asocia a hepatomegalia, esplenomegalia, nubosidad corneal y pérdida de audición. Tiene tres subtipos: congénito o hidrópico que inicia dentro del útero, infantil con inicio entre el nacimiento y los 12 meses, y juvenil con inicio después de los 2 años. La forma congénita presenta hydrops fetalis severo no inmune al nacer y ascitis. Los de presentación juvenil desarrollan puntos rojo cereza maculares y mioclonías 8,20.
Diagnóstico: Se basa en el aumento de excreción de ácido siálico en la orina y confirmado por el análisis genético de la deficiencia de neuroaminidasa en fibroblastos cultivados. El diagnóstico diferencial de la sialidosis tipo 1 es con otros tipos de EMP y de la sialidosis tipo 2 son otras enfermedades de almacenamiento similares 1,2,4.
Manejo: El antiepiléptico de primera línea es valproato, para tratar las mioclonías severas suele requerirse dos o tres drogas adicionales, que pueden ser benzodiazepinas, levetiracetam, zonisamida o topiramato. No se ha probado todavía el tratamiento de reemplazo enzimático en humanos, en modelos animales, no cruzaba la barrera hematoencefálica 20. Pronóstico: La esperanza de vida es muy corta, muerte temprana 4.
Gangliosidosis GM2 (enfermedad de Tay-Sachs y Sandhoff)
Grupo de enfermedades autosómicas recesivas caracterizadas por acumulación del gangliósido GM2, un tipo de glucolípido, en células neuronales 1.
Genética: Es de herencia autosómica recesiva, causado por mutación en los genes HEXA (enfermedad de Tay Sachs y variantes), HEXB (enfermedad de Sandhoff y variantes) y GM2A (variantes AB de la gangliosidosis GM2). Las proteínas codificadas por estos 3 genes son las subunidades α (gen HEXA), β (gen HEXB) de la enzima β-hexosaminidasa A. La tercera es una pequeña proteína de transporte de glucolípidos, la proteína activadora de GM2 (GM2A), que actúa como un sustrato cofactor específico para la enzima. La deficiencia de alguna de estas proteínas lleva a la acumulación de gangliósido, principalmente en lisosomas de células neuronales, causando muerte neuronal 1.
Epidemiología: La frecuencia de la enfermedad de Tay Sachs es 1 en 300 personas de la población general, siendo más común en ciertas poblaciones como quebequenses orientales en Canadá (frecuencia 1 en 14) y judíos asquenazis (frecuencia 1 en 30) 21. La enfermedad de Sandhoff tiene una incidencia de 1 en 1 000 000 en la población general 19.
Clínica: Tiene 3 subtipos clínicamente indistinguibles, más allá de ligeras manifestaciones viscerales y esqueléticas en algunas personas con enfermedad de Sandhoff. La primera forma descubierta fue la enfermedad de Tay-Sachs y sus variantes (gangliosidosis GM2 tipo I), la segunda fue la enfermedad de Sandhoff y variantes (gangliosidosis GM2 tipo II) y la tercera, la deficiencia de activador GM2 (gangliosidosis GM2 tipo III) (21,22). Los pacientes afectados por enfermedad de Tay-Sachs son normales al nacer, en el primer año de vida desarrollan deterioro psicomotor rápidamente progresivo, crisis epilépticas, hipotonía, ceguera, demencia y muerte 1,22. Las variaciones raras se inician más tarde en la vida y tiene una degeneración neurológica más lenta, presentándose primero con problemas en el balance y dificultad subiendo escaleras, viéndose un patrón característico de debilidad a nivel de músculos tríceps, iliopsoas y cuádriceps 21,22.
La enfermedad de Sandhoff-Jatzkewitz tiene una variedad de fenotipos. Suele iniciar a los 3-6 meses. Presentan retraso mental y motor, apariencia facial parecida a una muñeca, puntos color cereza en la retina, respuesta alarmada a los sonidos, macrocefalia, crisis epilépticas, ceguera temprana e hipotonía. Hay formas de inicio tardío que tienen supervivencia prolongada (20). La gangliosidosis GM2 de inicio tardío muestra una amplia variedad de síntomas, incluyendo ataxia cerebelosa, distonía, enfermedad de motoneurona, síntomas psiquiátricos, demencia y raras veces polineuropatía 1.
Manejo: No existe actualmente un tratamiento específico para esta enfermedad, solo se da tratamiento sintomático como antiepilépticos para control de crisis 21.
Diagnóstico: En casos sospechosos, de infantes con enfermedad neurodegenerativa con arresto del desarrollo y características que indican enfermedad cerebral y cerebelosa, se deben hacer pruebas de actividad enzimática de β-hexosaminidasa, y también análisis genético que identifiquen variantes patogénicas y probablemente patogénicas en los genes HEXA y HEXB 21.
Pronóstico: La muerte suele pasar antes de los 4 años de edad. En la enfermedad de Tay-Sachs ocurre a los 3-5 años, y en la enfermedad de Sandhoff a los 2-5 años 21,22.
Enfermedad de Gaucher
Es la enfermedad de acumulación lisosomal más común, causada por deficiencia de glucocerebrosidasa 1.
Genética: Herencia autosómica recesiva. Variantes en el gen GBA, que codifica la enzima glucocerebrosidasa (ácido β-glucosidasa), que convierte glucocerebrosida en ceramida y glucosa. Por esta alteración, la glucocerebrosida en el sistema reticuloendotelial. Se han descrito más de 400 variantes patogénticas y probablemente patogénicas causales de Gaucher1,22.
Epidemiología: La enfermedad de Gaucher tipo 1 tienen una frecuencia de 1 en 855 en judíos asquenazis. El tipo 2 tiene una incidencia de entre 1 en 100 000 y 1 en 500 000 nacimientos 22.
Clínica: Hay 3 tipos clínicos de enfermedad de Gaucher, que reflejan el grado de deficiencia de β-glucocerebrosidasa. Tienen en común hepatoesplenomegalia, lesiones óseas y compromiso ocasional de pulmones y otros órganos. El tipo 1 no tiene compromiso del sistema nervioso central. Los fenotipos de tipo 2 y 3 pueden ser difíciles de diferenciar 3,22.
El tipo 2, la forma aguda neuropática, tiene un inicio temprano con compromiso severo del sistema nervioso central y muerte temprana. Inicia entre el nacimiento y los 6 meses de vida. Su clínica incluye estrabismo, apraxia oculomotora, trismus, disfagia, retroflexión de la cabeza, pulgares corticales, rigidez de extremidades, aumento de reflejos osteotendinosos profundos, signo de Babinski positivo, infecciones pulmonares y falla en el crecimiento. Algunos tienen crisis epilépticas. La presencia de hepatoesplenomegalia en un infante con anormalidades oculomotoras y retroflexión de cabeza debe hacer sospechar de enfermedad de Gaucher tipo 2 22.
El tipo 3, la forma subaguda neuropática, tiene síntomas neurológicos con inicio tardío y curso más crónico que el tipo 2. La edad media de inicio es 1 año. Las manifestaciones más comunes son enlentecimiento y defectos en los movimientos sacádicos horizontales de los ojos. Algunos pueden desarrollar ataxia, espasticidad, acinesia, y crisis mioclónicas, con grados variables de demencia. Hay una variante llamada tipo 3c que presenta alteraciones cardiovasculares (calcificaciones en válvula aórtica, válvula mitral, y aorta ascendente). Se puede acompañar de nubosidad corneal, hidrocefalia y oftalmoplejía supranuclear 3,22.
Diagnóstico: Se debe sospechar en pacientes con esplenomegalia no explicada. Se realiza con deficiencia de actividad de glucocerebrosidasa en leucocitos, o fibroblastos. Estudio genético que identifique variantes en homocigosis o heterocigosis compuesta en el gen GBA. Existen variantes particularmente frecuentes en población con ascendencia judia asquenazi 22.
Manejo: El manejo se divide en terapia dirigida al defecto básico y cuidados de soporte. La terapia dirigida incluye el reemplazo enzimático, reducción del sustrato con miglustat o eliglustat y trasplante de médula ósea. Estos tratamientos proveen mejora en todos los síntomas excepto los neurológicos. Los cuidados de soporte incluyen tratamiento de osteomielitis si se desarrolla, reemplazos ortopédicos de cadera, analgesia para dolor óseo, esplenectomía en trombocitopenia severa y anemia, suplementación de bifosfonatos y vitamina D 22.
Pronóstico: Los pacientes con enfermedad de Gaucher tipo 2 fallecen en los primeros dos años de vida 22.
CONCLUSIONES
Hemos podido observar que las epilepsias mioclónicas progresivas son un grupo de enfermedades genéticas heterogéneas raras con punto en común la presentación de mioclonías de acción, crisis epilépticas y deterioro neurológico, que suelen cursar con un mal pronóstico en la mayoría de casos. Aunque se están haciendo avances hacia terapias genéticas específicas, todavía queda un camino por recorrer para conseguir tratamientos efectivos que mejoren la calidad de vida de los pacientes, y es ahí hacia donde se deben volcar esfuerzos de investigación a futuro. Hasta conseguir este objetivo, la mejor opción que tenemos es el tratamiento sintomático que no suele ser efectivo.

