











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Inteligencia
Inteligencia proviene del latín intelligentia, derivado del verbo intelligĕre y compuesta por intus (“entre”) y legere (“escoger”). Etimológicamente inteligencia hace referencia a quien “sabe elegir” 1.
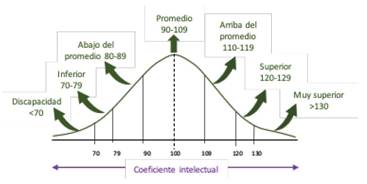
La inteligencia está definida como la capacidad para aprender de la experiencia y adaptarse, dar forma y seleccionar entornos 2, o como la capacidad para planificar, aprender “rápido”, razonar y resolver problemas 3,4; desempeñando un papel vital en el logro educativo, el éxito profesional y los “desenlaces” positivos o negativos en salud. 3,5 Se mide a través del coeficiente intelectual (CI) mediante diferentes pruebas (ej. Stanford- Binet, Wechsler), el cual varía a través de la vida y las generaciones (2). El CI tiene una distribución normal, donde la mayoría de las personas obtienen puntajes que se distribuyen en la parte central, clasificándolo en 7 grupos (gráfico 1) 6-9. Éste CI es variable a través de las diferentes poblaciones existiendo naciones que están alrededor de 70 y otras en 107, los cuales, además, se correlacionan fuertemente con los logros nacionales que se realizan en matemáticas y ciencias (ej.: TIMSS, PISA) 10,11. Según las teorías psicológicas como la teoría CHC, de inteligencias múltiples de Gardner y la triárquica de Stenberg, la inteligencia está basada en subestructuras fundamentales 2.
Se han descrito la relación que existe entre la inteligencia y cambios biológicos como: el volumen cerebral, grosor de la corteza cerebral y de regiones subcorticales, funcionamiento regional diferenciado, metabolismo de glucosa, neuroplasticidad, neuronas piramidales y el grosor de la sustancia blanca (Figura 1) 4. El grosor de la corteza cerebral, específicamente de los lóbulos frontal y temporal se ha correlacionado con la inteligencia. Es así que en los varones existiría una mayor asociación entre las cortezas prefrontal y temporal; mientras que en las mujeres la asociación sería entre las cortezas temporo-occipital 4. En ese mismo sentido, las personas con un coeficiente intelectual alto presentan niveles altos de funcionamiento en la corteza parietal, temporal y occipital, así como en la región subcortical del ganglio striatum, cerebros más voluminosos, mayor tamaño del fascículo uncinado derecho y un metabolismo de la glucosa más bajo durante los procesos de resolución de problemas 4,12,13. La relación entre inteligencia y el volumen de la corteza cerebral va cambiando durante la vida, como producto de una sobreproducción de sinapsis en la niñez y un aumento en la “poda” sináptica en la adolescencia-adultez 4. Además, se reconoce la importancia entre el número de las neuronas piramidales y sus conexiones dendríticas 4.
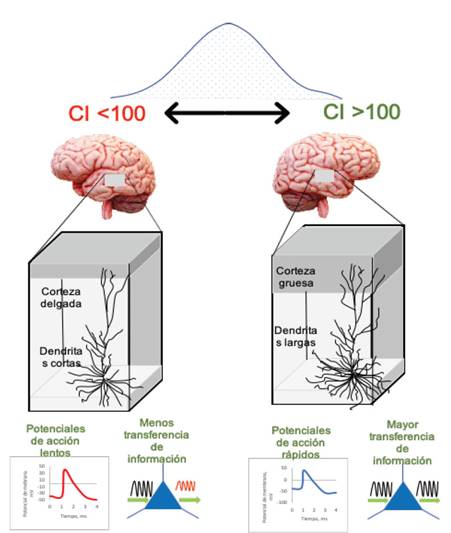
Figura 1 Bases celulares de la inteligencia humana. Un CI alto está relacionado con dendritas más largas, potenciales de acción más rápidos, y una transferencia de información a través de las células piramidales más rápido. Fuente: Extraído y traducido de Gourinova & Mansvelder (2019).
A pesar del gran avance en la genómica, no existe un gen o genes “claves” para el desempeño normal de la inteligencia. La inteligencia es un rasgo poligénico, donde cada uno de los genes aportan aproximadamente menos del 0,1% 2,4. Para poder identificar estos genes se ha utilizado la técnica de GWAS (del inglés genome wide association study) que utiliza las variantes únicas polimórficas o SNP (del inglés single nucleotide polymorphism)14). A la fecha, se han llegado a identificar a 1 041 genes, los cuales están involucrados en varios procesos como la neurogénesis, regulación del desarrollo del sistema nervioso central (SNC), regulación del desarrollo celular, proyección neuronal, diferenciación del SNC, sinapsis, diferenciación neuronal y de los oligodendrocitos e interacción célula-célula 4,15. Estos SNPs están localizados principalmente en regiones no codificantes, observándose que sólo el 1,4% de estos SNPs se encuentran en regiones exónicas y que la mayoría de estos están activos durante la embriogénesis (ej. DDX27, GNL3, NCAPG) 4. El cromosoma X contiene un gran número de genes (aproximadamente unos 120) que intervienen en el aprendizaje y memoria; y además, tiene una mayor densidad de estos genes con relación a los cromosomas autosómicos 16,17.
La heredabilidad se define como la proporción de la variación de los caracteres biológicos de una población, que es atribuible a la variación genotípica entre individuos y mientras más se acerque a uno, la influencia genética es mayor. La heredabilidad de la inteligencia es variable según las etapas del desarrollo encontrándose que podría estar en la niñez en 0,45 y en la adultez entre 0,80 a 0,86 18,19, mostrando que, al completar la maduración biológica, la inteligencia está influenciada fuertemente por la genética 20,21. Es más, se ha observado que piezas importantes de la inteligencia se desarrollan en la edad escolar 21. Una de ellas es el control cognitivo que es la capacidad de coordinar pensamientos y acciones para la realización de comportamientos dirigidos a objetivos, sirve como un proceso fundamental en funciones ejecutivas superiores, como el control ejecutivo de la atención, actualización (es decir, memoria de trabajo), desplazamiento e inhibición de la respuesta; este control cognitivo es fundamental en la cognición humana de nivel superior, como la inteligencia 21. Esta capacidad de control cognitivo es altamente hereditaria y está asociado a niveles altos de cognición, y se ha observado que esta heredabilidad varía desde la niñez (6-11 años) hasta la adolescencia (12-18 años) de 0,64 a 0,94 respectivamente 21.
Discapacidad intelectual
La discapacidad intelectual (DI) o conocida anteriormente como “retraso mental” es uno de los trastornos del neurodesarrollo más frecuentes 22,23, que se caracteriza por la limitación en el funcionamiento intelectual, así como dificultades conceptuales, sociales y áreas prácticas de la vida; que se observan en niños mayores de cinco años (o que pueda realizar la prueba correspondiente) y menores de 18 años 22,24. El diagnóstico de discapacidad intelectual requiere la presencia de los tres criterios siguientes 22,24:
Deficiencia en el funcionamiento intelectual, que incluye razonamiento, resolución de problemas, planeamiento, pensamiento abstracto, juicio, aprendizaje académico, aprendizaje desde la experiencia; el cual ha sido confirmado clínicamente de manera individualizada con pruebas estandarizadas de CI.
Déficit en el funcionamiento adaptativo que obstaculiza significativamente la conformidad con los estándares de desarrollo y socioculturales para la independencia y la capacidad del individuo de cumplir con su responsabilidad social.
La edad de inicio de estas deficiencias es durante la niñez.
La clasificación de la severidad de la discapacidad intelectual es en cuatro grupos (Tabla 1) 22.
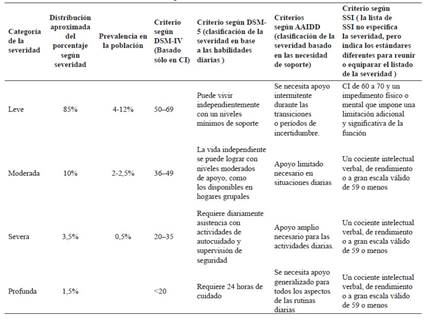
La prevalencia mundial de la discapacidad intelectual (DI) oscila entre 1 y el 3% de la población y el 90% tiene una etiología genética 25. Según la gravedad, la prevalencia variará (Tabla 1) y la relación varón: mujer es de 2:1 26,27. En Latinoamérica la prevalencia de DI varía entre el 3% al 12,9% de la población 28, sin embargo, estos valores podrían ser más altos llegando al 18,3% en países de bajos o medianos ingresos 29-31. En el Perú no se tiene registro sobre la prevalencia de DI, sin embargo, se estima que entre 0,9 y 5 millones de peruanos tendrían DI.
La DI tiene un impacto social y económico muy alto en la salud pública; es así, que los costos durante la vida de un paciente con DI (en EE. UU. y Europa) está entre 1-2 millones de dólares, siendo más costoso que la demencia y el cáncer 31.
Etiología de la discapacidad intelectual
La DI, así como el retraso del desarrollo psicomotor (RDPM) y el trastorno del espectro autista (TEA) son síntomas y signos que no expresan la etiología subyacente 32. El RDPM se define como el retraso en niños menores de cinco años en dos o más de las siguientes áreas: i) motora (gruesa y fina), ii) habla y lenguaje (expresivo y receptivo) iii) social y emocional y iv) cognitivo 23. Y el TEA describe un comportamiento caracterizado por incapacidad en la interacción social, restricción de intereses y comportamientos repetitivos, cuyo diagnóstico se realiza a través de pruebas neuropsicológicas especializadas 33-35. Estos tres trastornos del neurodesarrollo tienen que ser diferenciados y la DI se puede o no manifestar en la infancia como un retraso del desarrollo psicomotor, y pueden o no asociarse a TEA. Por lo tanto, todo paciente que presente alguna de estas características se deberá buscar la etiología.
La DI se clasifica en sindrómica si está asociada a una patología específica (ej. anomalías congénitas, talla baja, obesidad, epilepsia) y en no sindrómica o aislada 36.
La etiología de la DI es muy heterogénea, es así como una forma práctica de clasificarlas es de la siguiente manera: i) genéticas ii) teratogénicas iii) otras (Figura 2) 32.
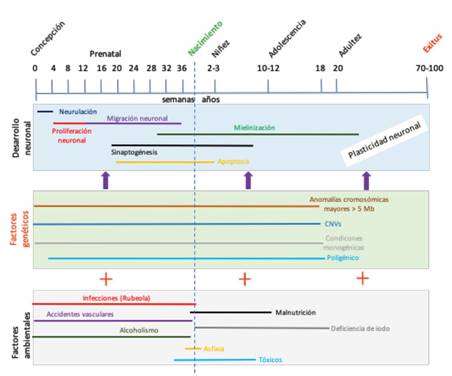
Figura 2 Etiología de la discapacidad intelectual. Los trastornos genéticos y ambientales pueden influir negativamente en el desarrollo neuronal. Estas variantes genéticas y ambientales actúan en conjunto para provocar un determinado fenotipo, el cual se puede observar, a través de otras comorbilidades, desde la etapa prenatal (ej. Malformaciones), neonatal (ej. Hipotonía, epilepsia), niñez (ej. Retraso del desarrollo psicomotor, TEA) o en la etapa adulta (ej. Esquizofrenia). Fuente: Extraído, traducido y modificado de Le Hellard & Steen (2014).
Etiología genética
La discapacidad intelectual tiene un origen genético hasta en un 90% de los casos estudiados, utilizando toda la tecnología disponible hasta el momento 25,37,38.
Las enfermedades genéticas que presentan DI aislada o sindrómica son causadas por variantes de un único nucleótido o SNV (del inglés single nucleotide variation), de múltiples nucleótidos o MNV (del inglés multinucleotide variation) o las variantes en el número de copias o CNV (del inglés copy number variation) 39. Estas variantes genéticas pueden ser determinantes para modificar la inteligencia, la cual dependerá de su función, la frecuencia de aparición con relación a la población y su interrelación con el medio ambiente (Figura 2) 36.
En pacientes con DI no sindrómica, se ha descrito que existe una alteración en la neurotransmisión glutamatérgica y dopaminérgica 40. Estas alteraciones excitatorias-inhibitorias no sólo ocurren en el cerebro, sino también en el conectoma cerebelar (cerebelo-núcleo-tálamo) 41.
Enfermedades monogénicas
Provocado por variantes en un solo gen y pueden ser SNV, MNV o CNV 39.
En OMIM (del inglés online mendelian inheritance in man) existen más de 708 entidades que presentan discapacidad intelectual (ver: www. omim.org). Sin embargo, aún existen entidades que a la fecha no han sido catalogadas en OMIM (gráfico 2); describiéndose a más de 1000 loci asociados a discapacidad intelectual 42, y que el número de genes implicados serían más de 2065 genes, según el portal de SysID (http://sysid.cmbi.umcn.nl/) 43,44. Los tipos de herencia son enfermedades dominantes autosómicas (ej. síndrome Sotos), dominante ligado al cromosoma X (ej. síndrome del X frágil), recesivas autosómicas (ej. errores innatos del metabolismo), recesivas ligadas al cromosoma X (ej. síndrome de deficiencia de creatina 1) o mitocondriales.
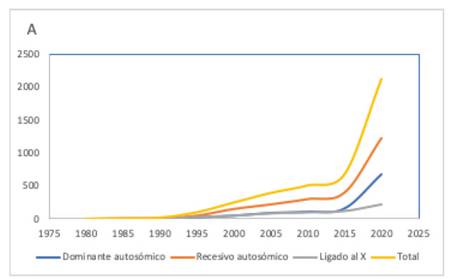
Gráfico 2 Número de genes descritos anualmente que están ligados a DI aislada y DI sindrómicas, observándose un crecimiento exponencial en estos últimos años. Fuente: Extraído, traducido y modificado de Vissers et al. 2017
Existe un mayor número de genes que están relacionados a la herencia recesiva autosómica, en comparación a los otros tipos (gráfico 3). Se ha reportado, que el número de genes que con certeza provocan DI de herencia recesiva autosómica estaría entre 1 222- 2 000 44,45, y los genes candidatos adicionales serían de 1 127 44.
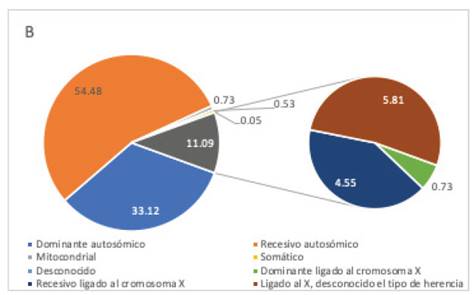
Gráfico 3 Proporción de genes según el tipo y patrón de herencia. Existen más genes de herencia recesiva autosómica. Fuente: Elaboración propia, datos extraídos de http://sysid.cmbi.umcn.nl/.
En las entidades de herencia dominante, éstas pueden ser por variantes de novo, heredadas de uno de los padres o mosaicismos gonadales 46. Lo que se conoce es que un neonato presenta 50-100 variantes de novo a nivel diploide (ej. 38 variantes por sustitución, 3 pequeñas inserciones-deleciones y una variante en splicing), resultando en 0,86 variantes que modifican un aminoácido 42,47.
Como se mencionó la discapacidad intelectual es más frecuente en varones que en mujeres (2:1).
Esta diferencia se debe a la presencia de un segundo cromosoma X en la mujer y a la cantidad y densidad de genes relacionados con la inteligencia que contiene este cromosoma 16,17.
Alteraciones cromosómicas
En los pacientes que tienen DI profunda, se puede observar anomalías cromosómicas microscópicas en el 12-35% de los casos, en DI moderada-severa ~3%, y en los casos leves entre el 1-3% (promedio del 4,6%) 26,31.
Dentro de las alteraciones numéricas o aneuploidías, se tiene como ejemplo al síndrome Down, con una frecuencia de 3,45/700 recién nacidos vivos 48, y corresponde al 12%-36% de los casos con DI 31,49, donde la fisiopatología de la DI es por triplosensibilidad o sobrexpresión en varios genes (ej. DYRK1A, RCAN1, RIP140, CSB, miR155, SOD1, APP) que están involucrados en procesos del neurodesarrollo (ej. neurogénesis, plasticidad sináptica, neurotransmisión) o aumento en la neurodegeneración (ej. estrés oxidativo, patología amiloide o muerte celular neuronal)50).
Las variantes en el número de copias o CNV (del inglés copy number variation) o conocidas anteriormente como microdeleciones - microduplicaciones, son segmentos de ADN mayor a 1 kb 51. Las CNVs más frecuentes tienen una prevalencia entre 1/1 000 a 1/25 000 52; y los fetos tienen una incidencia de 0,7%53. En los pacientes con DI se pueden encontrar como causa de la DI en el 20-25,8% de los pacientes 54. Dependiendo de si existiera una CNV en ganancia o pérdida provocará triplosensibilidad o haploinsuficiencia respectivamente. Otra manera de provocar un fenotipo es por la fusión de genes o disrupción de estos. Se ha encontrado que algunas CNVs que no contienen genes (CNVs no codificantes), son regiones reguladoras de otro(s) gen(es) 39,55,56,57. Las alteraciones subteloméricas (un tipo de CNVs), provocan entre el 5-7% de los pacientes 58. Otras CNVs frecuentes son por ejemplo el síndrome Prader-Willi/Angelman, síndrome Williams, síndrome de deleción 22q11.2 31.
Etiología teratogénica
Existen diversos teratógenos que tienen efectos adversos en el desarrollo del sistema nervioso central 59. El mecanismo de sus efectos es muy complejo y depende de diversos factores como del grado y tiempo de exposición, de la susceptibilidad genética de la madre y el feto, del tipo de teratógeno, entre otros. Se ha demostrado que la exposición aguda o crónica a factores ambientales como el abuso de drogas o sustancias tóxicas durante períodos críticos del desarrollo causa alteraciones globales o específicas de genes a través de modificaciones de histonas, remodelación de cromatina o metilación de ADN en diferentes áreas del cerebro 60.
Entre estos factores ambientales que actúan como teratógenos en el desarrollo cerebral tenemos al alcohol, observándose que el consumo aún en cantidades mínimas, tienen un riesgo de tener hijos con trastorno del espectro alcohol fetal, provocando cambios epigenéticos que modularían la expresión y función génica, y que además podría influir en generaciones posteriores.61-64) Específicamente, se postula que durante la exposición al alcohol en la etapa embrionaria-fetal el mir-9 disminuye, quien está relacionado en la segmentación cerebral, neurogénesis y maduración neuronal 65. Otros teratógenos, relativamente frecuentes, se tiene a la isotretinoina 66,67 (induce la expresión anómala de genes implicados en la diferenciación neuronal) 68, al misoprostol 69,70, la deficiencia de ácido fólico 71 y la infección por rubeola 72,73.
Factores de riesgo
Los factores de riesgo de este trastorno del neurodesarrollo son consanguinidad, la edad materna mayor a 35 años, educación materna menor a 12 años, pretérmino (<37 semanas), sexo masculino, bajo peso al nacer (<2 500), paridad (>= 3), alcoholismo, tabaquismo y epilepsia y asma materno 74,75.
El riesgo de aparición de variantes en el número de copias de novo está relacionado a madres mayores de 35 años 76. Sin embargo, es importante precisar que la edad paterna mayor a 29,7 años es un factor de riesgo para la aparición de variantes de novo (sean SNV, MNV o CNV) asociados a trastornos del neurodesarrollo, incluida la DI 77.
Prevención de la discapacidad intelectual
Están incluidas medidas como el tamizaje neonatal universal para condiciones potencialmente tratables (ej. hipotiroidismo congénito, fenilcetonuria), vacunación contra rubeola, administración de ácido fólico, mejoras en el cuidado del embarazo, evitar el consumo de alcohol 78,79. Específicamente los errores innatos del metabolismo tienen tratamiento refinados, de ahí la importancia de su diagnóstico oportuno a través del tamizaje neonatal universal 29. Además, es importante el disminuir las uniones matrimoniales consanguineas.
Comorbilidades
La discapacidad intelectual puede acompañarse en el 30-57% con otros trastornos del neurodesarrollo, como encefalopatía epiléptica, TEA, esquizofrenia, parálisis cerebral, ceguera e hipoacusia 22,43,80. Otras comorbilidades son trastorno bipolar, trastorno de hiperactividad y déficit de atención 59.
Diagnóstico etiológico de la discapacidad intelectual.
El diagnóstico de DI nos permite: i) conocer la etiología, ii) pronóstico o conocer el curso clínico, iii) informar a la familia sobre los mecanismo(s) genético(s) y el riesgos de recurrencia familiar, iv) optar tratamientos refinados, v) racionalizar las pruebas de diagnóstico, vi) proveer información relativa al tratamiento, los síntomas, gestión o vigilancia de complicaciones, vii) prestación de apoyo a la familia según la condición específica, viii) acceso a los protocolos de tratamiento de investigación, y ix) garantizar una mejor salud, así como una atención sociosanitaria, donde exista una satisfacción de los servicios, interpretados de una manera adecuada según los resultados (niño y su familia) 81.
El diagnóstico clínico de la etiología de la discapacidad intelectual se realiza utilizando la siguiente estructura (Figura 3):
i. Antecedentes prenatales, natales y del desarrollo psicomotor.
ii. El pedigrí de tres generaciones, el cual ayuda a precisar consanguinidad, antecedentes familiares con patología similar o psiquiátrica (ej. psicosis, depresión, TEA) 32.
iii. El examen físico incluyendo una antropometría detallada, evaluación de dismorfias, uso de lámpara de Wood y el registro fotográfico (con la finalidad de realizar comparaciones con casos similares anteriores o compartir la información con otros especialistas) 32,82,83.
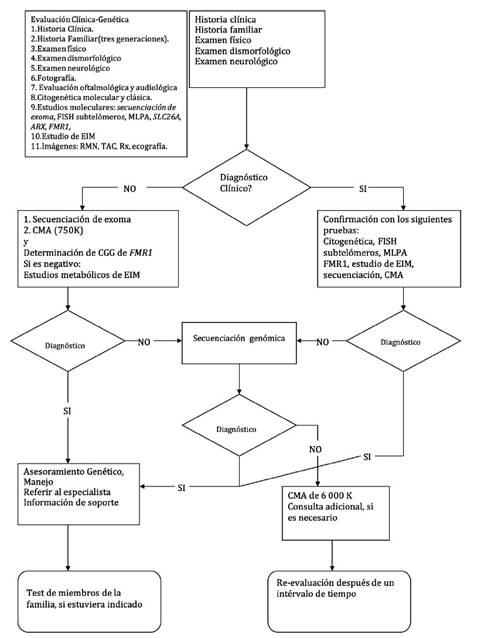
Figura 3 Algoritmo diagnóstico de la discapacidad intelectual. EIM= Errores innatos del metabolismo, CMA= Análisis cromosómico por micromatrices, MLPA= Multiple ligation probe amplification. Fuente: Elaboración propia.
Se ha determinado que los datos de la anamnesis general, antecedentes prenatales, perinatales, familiares y una exploración dismorfológica adecuada orientan certeramente el diagnóstico entre el 39% y 81% de los casos 82,83. La exploración neurológica contribuye hasta en el 42% al diagnóstico sindrómico 82,83. El uso de motores informático de búsqueda de síndromes específicos síntomas o dismorfias faciales específicas como OMIM (www.omim.org), Face2gene/LMD (London Medical Databases), Possum, Phenomizer, Findzebra; contribuyen en el diagnóstico diferencial 84-92.
Si existe una sospecha clínica alta debido a que el paciente presenta características “patognomónicas” de alguna enfermedad genética, es siempre mejor realizar la prueba genética que dará la confirmación; el cual podrá ser cariotipo, test de metilación, análisis cromosómico por micromatrices, o secuenciación. Por ejemplo, en pacientes con distonía y/o epilepsia se puede determinar las variantes en el gen ARX 93. En cualquier varón con hipotonía congénita y un retraso del desarrollo psicomotor severo se deberá examinar la presencia de variantes del gen MECP2 94. Entre el 0,2-3,5% de los varones con DI, se ha observado variantes del SLC6A8, el cual provoca el síndrome de deficiencia de creatina cerebral 95. En ese mismo sentido, si existiera la sospecha de alguna CNV específica (ej. síndrome Williams, síndrome de deleción 22q11, síndrome Prader-Willi), se puede realizar FISH (del ingles fluoroscence in situ hibridization) o MLPA (del inglés múltiple ligation probe amplification) 96. Las pruebas genéticas son complementarias entre sí, ya que a la fecha no se ha identificado una prueba que pueda detectar de manera simultánea todas las variantes genéticas, llámese SNV, MNV o CNV.
El cariotipo (mayor a 650 bandas) detecta la etiología sólo en el 3% de los pacientes con DI sin un diagnóstico clínico específico 82. Debido a que las variantes en el número de copias mayores a 3-5 Mb, es detectada si la resolución es de 1 000 bandas, y CNVs de 10 Mb si la resolución es de ~400-500 bandas 97.
En aquellos pacientes sin un diagnóstico clínico, se procederá en realizar exámenes que evalué todo el genoma. Así, comenzaremos con la secuenciación de exoma/genoma; luego se utilizará el análisis cromosómico por micromatrices o CMA (del inglés chromosomal microarray analysis).
La secuenciación masiva, específicamente la secuenciación de exoma tiene una tasa de detección superior al CMA 98 y que está alrededor del 31% y 53% en pacientes con DI aislado y sindrómico, respectivamente (promedio del 36%) 99, por lo que actualmente debería ser la primera prueba de elección para determinar la etiología de la DI 100.
El CMA sirve para determinar CNVs (deleciones o duplicaciones submicroscópicas) el cual en algunos centros sigue siendo el primer test de elección en niños con DI. 101,102 El uso de CMA en pacientes con DI detecta la etiología entre el 15-25,8% de los casos 101-103; sin embargo, si sólo se realiza CMA a los pacientes con DI y dismorfia facial el rango puede llegar entre el 33-40% 104-106. Además, las que utilizan marcadores SNP tienen la ventaja adicional de poder determinar regiones de homocigosidad o ROH (del inglés región of homozigosity) el cual si estuviera alterada indicaría posibles disomías uniparentales o incluso indicarnos coeficiente de endogamia altos, lo cual estaría en relación con una posible entidad de herencia recesiva autosómica 75. Las disomías uniparentales, según la región afectada, estaría relacionada a genes en los que se ha modificado la impronta o la aparición de enfermedades recesivas autosómicas 107.
Es importante precisar que todo paciente con RDPM/DI, y en especial en aquellos sin compromiso motor, debería verificarse la expansión de tripletes CGG del gen FMR1 (síndrome del X frágil). El síndrome del X frágil se observa en el 2-3% de los varones y en el 1-2% de las mujeres con DI 83, siendo así la segunda causa más frecuente de DI, después del síndrome Down 108.
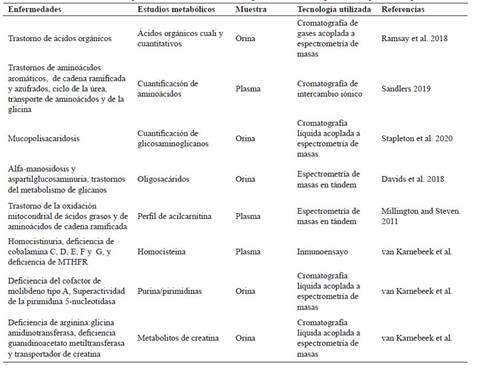
Las pruebas para determinar errores innatos del metabolismo son múltiples y nos pueden ayudar en el diagnóstico en un 0-8,4% de los pacientes con DI, con un promedio del 1% 106,109. Estos exámenes son generales (ej. lactato, gases venosos, electrolitos, perfiles hepático y tiroideo, cetonas, amonio, magnesio, ceruloplasmina, cobre, hierro, CPK) y los especializados (Tabla 2) 27,106,110. La ventaja de búsqueda de este grupo de patologías es que existen al menos 81 entidades que tienen una terapéutica específica (http://www.treatable-id.org)80,111,112) por lo tanto el cribado neonatal universal mediante políticas públicas es de suma importancia, con la finalidad de diagnosticar oportunamente y mejorar a largo plazo los objetivos del tratamiento 27.
Tabla 2 Estudios metabólicos especializados utilizando tecnología metabolómica, para la búsqueda de algunos EIM.
Otros exámenes auxiliares como la TEM, y la resonancia magnética (RMN) que permiten establecer las alteraciones estructurales del desarrollo cerebral ayudan en el diagnóstico etiológico entre el 0%-3,9%, siendo más sensible el uso de la RMN 109. En este mismo sentido, el uso del electroencefalograma, no tiene utilidad para fines de diagnóstico etiológico 109.
Es necesario destacar que existen una plétora de estudios donde se analiza sobre el costo/efectividad superior del CMA y de la secuenciación masiva para determinar la etiología de la DI, a pesar del coste elevado, en comparación a pruebas de rutina como la RMN, electroencefalograma o el propio cariotipo 98,113-118.
Es importante indicar que siendo la inteligencia y la discapacidad intelectual un rasgo continuo poligénico, se debería realizar una medición del CI en los padres y hermanos del paciente con discapacidad intelectual, con la finalidad de establecer los rangos del CI familiar, principalmente en aquellos niños- adolescentes que tienen un rendimiento escolar bajo pero que no necesariamente presentan un CI menor a 70, y a que a pesar de estar dentro de los rangos usuales, podría tener una condición genética, que eventualmente incluso podría ser de novo y heredado a su descendencia (gráfico 4). Por lo tanto, se podría utilizar la siguiente fórmula (CI +CI )/2±30, (donde CI = Coeficiente intelectual paterno y CI = Coeficiente intelectual materno). Lo anterior expuesto está basado en que los rasgos poligénicos familiares, son generalmente parecidos; y además se ha observado que en pacientes con DI leve, los hermanos suelen tener un CI por debajo de la curva normal, en comparación con aquellos con los pacientes con DI severa 119.
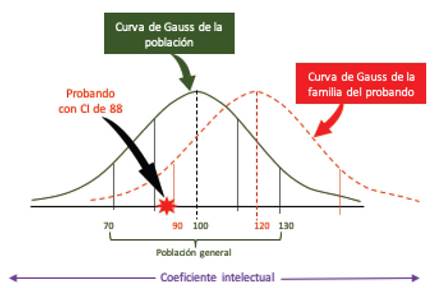
Gráfico 4 Medición del CI familiar (en el ideal padres, hermanos, tíos) con la finalidad de estimar la curva normal familiar, en pacientes con bajo rendimiento escolar y CI>70. Si la curva estimada “familiar” del CI estuviera por encima, sería necesario utilizar el protocolo de atención de pacientes con DI. En el ejemplo, se observa que el probando tiene un CI de 88; no obstante, a pesar de estar dentro de las curvas normales, está por debajo del estimado familiar. Fuente: Elaboración propia, los puntos de corte de la población general son extraídos de Wechsler (2008).
Manejo integral de la discapacidad intelectual
La heterogeneidad etiológica y clínica de la DI requiere por lo tanto consideraciones individualizadas; sin embargo, se puede tener un marco general para el manejo de los pacientes con DI, teniendo las siguientes consideraciones: i) tratamiento de complicaciones médicas; ii) atención médica preventiva general; iii) tratamiento de condiciones médicas y de otras comorbilidades neurológicas; iv) tratamiento de comportamientos agresivos; v) servicios de rehabilitación; vi) soporte educativo; vii) formación profesional; viii) apoyo social; ix) apoyo a la vida comunitaria; x) soporte para el nivel apropiado de empleo; xi) transición de servicios pediátricos a adultos; xii) tutela, consideraciones financieras y legales. El modelo óptimo de atención para personas con discapacidad intelectual es un modelo de atención interdisciplinaria en el marco de un servicio clínico o centro médico. El profesional médico de atención primaria debe brindar atención médica preventiva y general, además de facilitar y coordinar la atención consultiva especializada y el acceso a los servicios de apoyo necesarios 27.
Se están creando las estrategias necesarias para el descubrimiento de nuevas drogas, utilizando la reprogramación de las células madre y edición génica, con la finalidad de realizar una medicina personalizada 42,120.
CONCLUSIONES
La capacidad de determinar la etiología de la discapacidad intelectual es un reto constante para el especialista. La correcta determinación de la etiología resulta del criterio, experiencia y el seguimiento de las guías clínicas. La pericia clínica y el uso prudente de los recursos diagnósticos es vital en regiones con recursos limitaciones como Perú y varios otros latinoamericanos. La determinación de la etiología de la discapacidad intelectual permitirá el adecuado asesoramiento genético, conocer el pronóstico y el planteamiento de alternativas terapéuticas más específicas. Actualmente, se puede determinar la determinar la etiología de DI hasta en un 90% de los casos utilizando las nuevas tecnologías ómicas, como el análisis cromosómico por micromatrices y la secuenciación masiva.

