











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCION
La encefalitis de Rasmussen (ER) es una patología infrecuente ocasionada por inflamación crónica autoinmune, habitualmente de un solo hemisferio cerebral. Se caracteriza por presentar déficit neurológico focal progresivo, epilepsia focal farmacorresistente y deterioro cognitivo, asociados a hemiatrofia cerebral 1-3. Típicamente, suele iniciar en la infancia con una incidencia de hasta 2.4 casos por cada 10 millones de niños por año. Su inicio en la adolescencia o adultez es infrecuente, siendo esta variante conocida como encefalitis de Rasmussen de inicio tardío (ERIT) 1,2,4,5.
En niños, suele presentar una evolución dividida en tres etapas: Pródromo, etapa aguda y etapa residual. La primera fase se caracteriza por presentar crisis focales esporádicas con déficit focal leve o sin éste, con una duración promedio de 7 a 12 meses; tras ello, sigue una fase aguda, en la cual las crisis se tornan recurrentes, farmacorresistentes hasta progresar a epilepsia parcial continua (EPC) hasta en un 90% de los casos y se añade compromiso neurológico, que se manifiesta habitualmente como déficit focal (hemiparesia, afasia, hemianopsia) y deterioro cognitivo. En la fase residual los pacientes permanecen con el daño neurológico establecido y persisten las crisis epilépticas, aunque con menor frecuencia 1,2,4,5. Sin embargo, se ha reportado que los casos con ER de inicio tardío presentan características distintas y una progresión más lenta, por lo que podrían ser infradiagnosticados en sus etapas tempranas 1,2,4,6. Además de la dificultad diagnóstica de la ERIT, puede existir fracaso al tratamiento médico basado en inmunoterapia, siendo la alternativa quirúrgica una herramienta útil en el control de crisis farmacorresistente en casos discapacitantes 1-8.
En esta serie, presentamos tres casos de encefalitis de Rasmussen de inicio tardío, confirmados con histopatología e inmunohistoquímica, cuyo diagnóstico no fue sospechado desde el inicio. Describimos su evolución temporal hasta su diagnóstico definitivo y resultado post quirúrgico.
CASO 1
Paciente mujer, de 20 años, diestra, sin antecedentes de asfixia al nacer, crisis febriles, trauma craneoencefálico o infecciones del sistema nervioso central y con desarrollo psicomotor normal. A los 13 años inicia con crisis focales sin alteración de conciencia, oculocefaloversivas hacia la izquierda. En el curso de 3 años desarrolló farmacorresistencia (fenitoína 300mg/día y valproato 2g/día) y las crisis evolucionaron con un cambio en su semiología: aura de parestesia en miembro superior izquierdo, seguidas de clonías en hemicuerpo izquierdo y oculocefalogiria hacia la izquierda con preservación de la conciencia y el lenguaje. En el postictal siempre tenía hemiparesia izquierda transitoria. Gradualmente desarrolló discreta hemiparesia izquierda (fuerza muscular 4+/5), psicosis crónica y conducta pueril, con un incremento de la frecuencia de sus crisis hasta 4 a 7 veces por semana. La resonancia magnética (RM) de encéfalo evidenció leve engrosamiento cortical con discreta hiperseñal en región fronto-central derecha, sugerente de displasia cortical focal (DCF) además de mínima atrofia focal frontoparietal perilesional. A los 18 años, un primer video-electroencefalograma (video-EEG) mostró descargas epileptiformes interictales/ictales en zona frontocentral derecha. Se planteó entonces una posible zona epileptogénica (ZE) en región central dorsolateral derecha y se procedió a corticotomía central derecha guiada por electrocorticografía (ECoG). El estudio de anatomía patológica (AP) reportó gliosis. Tras ello, la paciente presentó mejoría significativa, con crisis similares muy esporádicas (1 vez cada 4 a 6 meses), sin modificar su esquema de fármacos anticrisis. Después de 3 años de la cirugía, tras COVID-19 moderado (neumonía leve sin insuficiencia respiratoria), presentó EPC con crisis sensorimotoras (parestesias y clonías) con progresión jacksoniana que iniciaban por el pie o la mano izquierdas y progresaban a todo el hemicuerpo ipsilateral. Para entonces se constató mayor hemiparesia izquierda (fuerza muscular 3-/5) y deterioro cognitivo. La paciente había llegado a ser tratada con 5 fármacos anticrisis a dosis máximas toleradas (fenitoína, levetiracetam, valproato, topiramato, clobazam) por más de 3 semanas y pulsos de corticoide, sin alguna mejoría. Se realizó una revisión de las imágenes de RM de encéfalo y se evidenció atrofia frontal progresiva y lesiones hiperintensas hemisféricas derechas evolutivas a predominio frontal no patentes en la primera RM (Figura 1(A)); un video-EEG de control mostró descargas epileptiformes inter ictales/ictales en amplia región central derecha con propagación eléctrica frontotemporal ipsilateral y frontocentral izquierda, planteándose entonces, ZE en amplia región pericentral derecha (Figura 1 (B)). Se planteó probable ER y se procedió a hemisferotomía funcional, siendo el hallazgo histopatológico: inflamación crónica perivascular y vacuolización de neurópilo; la inmunohistoquímica (IH) evidenció: CD3 positivo; CD20 positivo; CD68 microglía incrementada (Figura 1 (C)). Estos hallazgos confirmaron el diagnóstico de ER. Actualmente la paciente se encuentra en Engel Ia (completamente libre de crisis), con hemiparesia izquierda moderada (fuerza muscular 3+/5), que ha mejorado parcialmente con terapia física. La psicosis se encuentra controlada con risperidona.
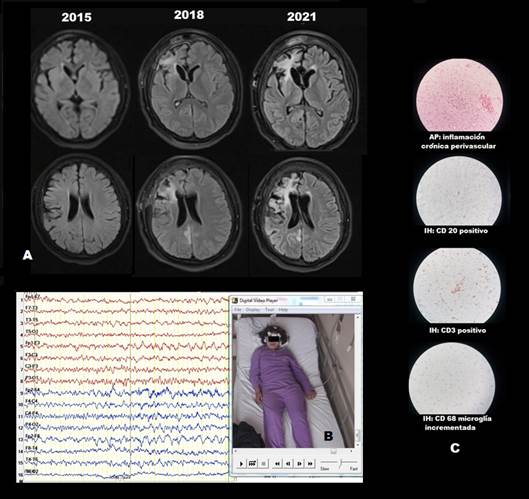
Figura 1 A: Evolución de RM de encéfalo (secuencia FLAIR) a lo largo de 6 años. Atrofia y lesiones hiperintensas derechas progresivas. B: Video-EEG actual. Registro ictal. Aura sensitiva en hemicuerpo izquierdo, seguida de clonias ipsilaterales. EEG ictal con inicio fronto-central derecho y propagación fronto-temporal ipsilateral y frontocentral izquierda.C: AP e IH. Hallazgos confirmatorios de ER.
CASO 2
Paciente mujer de 25 años, diestra; con antecedente de hemiparesia secuelar derecha leve a predominio braquial (fuerza muscular 4+/5), por probable evento vascular a los 10 años. Inició epilepsia focal a los 16 años, en contexto de gestación, con crisis diarias diurnas (hasta 4 veces/día) y de difícil control con fármacos desde su inicio; con aura de sensación de “cierre de la garganta con falta de aire” seguida con marcada hipersalivación, luego pérdida del contacto con el entorno asociada a postura distónica de miembro superior derecho, con duración aproximada de 1 minuto. A los 20 años, en base a un primer video-EEG (actividad epileptiforme ictal/interictal representada sobre región frontocentral izquierda), RM de encéfalo que mostró patología dual (esclerosis mesial temporal izquierda y lesión hiperintensa insular ipsilateral sospechosa de gliosis) y SPECT ictal con zona de hiperperfusión insular izquierda y en polo temporal izquierdo (Figura 2); se planteó ZE insulo-opercular y se realizó lobectomía temporal anteromesial izquierda (por patología dual) y corticotomía parcial insular ipsilateral. Los resultados de la AP no fueron concluyentes, aunque sugirió DCF y la paciente presentó mejoría significativa por 6 meses, con crisis residuales muy esporádicas (Engel IIb). Posteriormente se incrementó la frecuencia a 4 crisis semanales; manteniéndose la semiología, pero añadiéndose actividad hipercinética en extremidades derechas en el curso de las crisis y atenuándose la hipersalivación. Durante los últimos 4 años, las crisis se tornaron nuevamente esporádicas al optimizar los fármacos anticrisis en politerapia (carbamazepina 800mg/día, levetiracetam 3000mg/día, lamotrigina 350mg/día y clobazam 30mg/día). Sin embargo, en el 2021 la paciente desarrolló EPC incapacitante, con clonías faciobraquiales derechas, asociadas a marcada hipersalivación, que no mejoró con fármacos anticrisis más pulsos de corticoides y se evidenció incremento de la hemiparesia derecha (fuerza muscular 3+/5) y deterioro cognitivo leve asociado (determinado por combinación de baterías psicométricas de evaluación neuropsicológica). La RM de encéfalo evidenció lesión residual insular izquierda, pero además nueva lesión hiperintensa orbitofrontal y atrofia ipsilateral (Figura 2 (A) - RM de 2017). Un nuevo video-EEG evidenció actividad epileptiforme continua sobre región central izquierda con mayor electronegatividad en C3 (Figura 2 (B)). Fue intervenida nuevamente de lesionectomía insulo-opercular izquierda y desconexión fronto-insular. Luego de 4 meses de ésta última cirugía, la paciente se encuentra en Engel Ib, la EPC ha remitido, pero aún presenta breves (menores a 15 segundos) paroxismos de clonías peribucales derechas sin alteración de conciencia, interdiarios, que no le resultan incapacitantes (realiza actividades de la vida diaria y actividades instrumentales). Posterior a la cirugía, la hemiparesia derecha a predominio braquial se acentuó levemente (fuerza muscular 3/5) y no hubo compromiso del lenguaje. El estudio anatomo patológico informó parénquima cerebral con infiltrado linfoide perivascular con IH: CD3: positivo, CD20: positivo con escasas células, CD68: Microglías; hallazgos compatibles con ER (figura 2 (C)). Una RM de encéfalo reciente da cuenta de nuevas lesiones hiperintensas frontales dorsolaterales izquierdas, a pesar de que la mejoría clínica se mantiene (Figura 2 (A)).
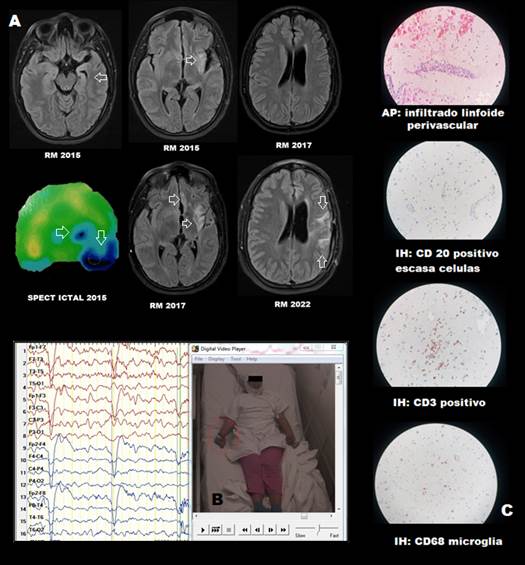
Figura 2 A: Neuroimágenes. RM de encéfalo 2015, patología dual. SPECT ictal con hiperperfusión insular y polo temporal izquierdos. RM 2017, lesión insular y orbitofrontal izquierda. RM de encéfalo 2022 lesión hiperdensa frontal dorsolateral izquierda. B: Video-EEG. Registro ictal temprano, con postura de miembro superior derecho posterior a aura de constricción laríngea. Actividad epileptiforme ictal temprana con mayor electronegatividad sobre región fronto-central izquierda. C: AP e IH. Hallazgos confirmatorios de ER
CASO 3
Paciente mujer de 45 años, diestra, sin antecedentes de asfixia al nacer, crisis febriles, trauma craneoencefálico e infecciones del sistema nervioso central; con desarrollo psicomotor normal y educación superior completa. A los 42 años, inició con crisis focales sin alteración de conciencia, sensorimotoras, con parestesias y clonías de progresión jacksoniana braquiofaciales derechas, asociadas después de meses a paroxismos de oculocefalogiria a la derecha, detención del habla y postura distónica braquial ipsilateral, con componente reflejo (exacerbación con el movimiento de la extremidad, con la alimentación, con emociones negativas y a veces con los ruidos súbitos e intensos). En los siguientes 3 años, las crisis se tornaron gradualmente interdiarias y resultaron muy incapacitantes, incluso para su alimentación. Desarrolló progresivamente paresia braquial distal derecha (fuerza muscular distal 3/5) y disminución del rendimiento intelectual en el campo laboral. Recibió valproato 2g/día, carbamazepina 1000mg/día y fenitoína 300mg/día, sin mejoría. Luego de ser catalogado su cuadro como distonía mioclónica en otro centro, la paciente suspendió los fármacos anticrisis. Entonces, el cuadro progresó a EPC e ingresó a nuestro centro. Las neuroimágenes de encéfalo evidenciaron angioma venoso parietal izquierdo y calcificación puntiforme subcortical precentral ipsilateral (Figura 3 (A)). El video-EEG prolongado sugirió ZE central dorsolateral izquierda con propagación a región frontal premotora ipsilateral. Se procedió a corticotomía central izquierda guiada por ECoG y mapeo de la corteza motora primaria mediante estimulación cortical (Figura 3 (C)). El hallazgo histopatológico fue de encefalitis a predominio linfocitario, con inmunohistoquímica: CD3 positivo predominante, CD20 positivo focal y CD68: macrófagos; hallazgos compatibles con ER (Figura 3 (D)). Actualmente, persiste con epilepsia parcial continua con solo leves clonías faciales derechas sin ser incapacitantes la mayoría del tiempo (realiza actividades de la vida diaria, actividades instrumentales y ha retomado sus labores), aunque muy esporádicamente se asocian a breve postura distónica braquial derecha (Engel II b). Tras la cirugía la paciente recibió pulsos de corticoides y de inmunoglobulina humana endovenosa (IgEV), además de recambio plasmático, sin obtener mejoría adicional.
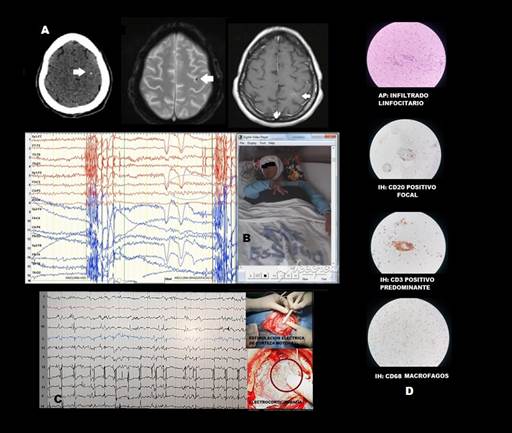
Figura 3 A.Neuroimágenes: TC y RM en T2 ecogradiente con calcificación puntiforme precentral izquierda y T1 con gadolineo con angioma venoso parietal izquierdo. B. Video-EEG. Registro ictal. Mioclonía braquiofacial derecha, evolución con postura distónica de miembro superior derecho y oculocefalogiria a la derecha. EEG con ritmos rápidos de bajo voltaje en región central izquierda previo a postura distónica. C: Electrocorticografía intraoperatoria con estimulación eléctrica de corteza motora primaria. D: AP e IH, hallazgos confirmatorios de ER.
DISCUSION
La encefalitis de Rasmussen es una enfermedad infrecuente, que suele afectar población pediátrica con una edad de presentación promedio de 6 años; aunque hasta un 10% de casos se reportan de inicio en adolescentes y adultos, considerándose encefalitis de Rasmussen de inicio tardío 1,2,5. Bien et al., en un consenso europeo, establecieron criterios diagnósticos en el 2005, considerándose dos combinaciones: Parte A, requiere 3 de 3 criterios diagnósticos: 1. Crisis focales (con o sin EPC) y déficit cortical unilateral; 2 EEG con lenificación hemisférica con o sin crisis de inicio unilateral/actividad epileptiforme unilateral; 3. Resonancia con atrofia cerebral cortical hemisférica asociada a hiperseñal en secuencias T2/FLAIR o atrofia/ hiperseñal de la cabeza del caudado ipsilateral. Parte B, con al menos 2 de 3 criterios: 1. Déficit neurológico cortical unilateral progresivo o EPC; 2. Resonancia con atrofia cortical hemisférica progresiva; 3. Histopatología que demuestre encefalitis dominada por linfocitos T, células microgliales activadas y astrogliosis reactiva 4.
Se ha reportado que los casos de ERIT podrían ser atípicos y variar en sus características y evolución comparado con la ER que inicia en la infancia: crisis epilépticas muy frecuentes desde el inicio e incluso farmacorresistentes, crisis sugerentes de un foco epileptógeno restringido, evolución mucho más lenta hacia la EPC, déficit neurológico focal más leve y de más paulatina instauración, infrecuente compromiso de regiones occipitales, menor grado o nula presencia de atrofia cerebral y menor o más sutil deterioro cognitivo 1,2,6,9-11. Una combinación de estas características las hemos evidenciado en nuestros tres casos y sumadas a su inicio en la adolescencia o adultez; las características atípicas o poco floridas en las neuroimágenes iniciales (ausencia de atrofia significativa y de hiperseñales en FLAIR y T2 de la RM) y resultados confusos o no concluyentes en la histopatología de las primeras cirugías de los casos 1 y 2; el diagnóstico inicialmente no fue sospechado. Olson et al, en el 2013 estimaron la sensibilidad de los criterios diagnósticos de Bien et al del 2005, principalmente por la descripción de casos atípicos en su presentación, encontrando una sensibilidad de 81%, especificidad de 92% y valor predictivo positivo de 77%. Describieron características atípicas para los criterios previos: resonancia con hiperseñal sin atrofia, ausencia de epilepsia o crisis generalizadas, EEG con lentificación o actividad epileptiforme bilateral, curso lentamente progresivo, presencia de hemicoreoatetosis y coexistencia de otras patologías con la ER, como displasia cortical focal. Por lo que propusieron ampliar el panel diagnóstico a aquellos pacientes que cumplieran 2 de los 3 criterios de la parte A asociados al criterio B3 (histopatología positiva). Su propuesta se hace más relevante en casos de ERIT como los que reportamos, en quienes es más recuente la presentación atípica 12.
Nuestras 3 pacientes presentaron control inicial solo parcial de las crisis epilépticas, con disminución en su frecuencia diaria o semanal tras medicación anticrisis; pero gradualmente fueron tornándose farmacorresistentes y evolucionaron a EPC en un periodo de 3 a 5 años. En niños se ha descrito una evolución más rápida en el establecimiento de las tres clásicas fases: prodrómica, aguda y residual, llegando a una fase residual en 1 a 2 años desde el inicio de las crisis (1-4, 8, 13).
En nuestra serie, las crisis tenían un aparente foco epileptógeno único y restringido en los estudios iniciales, lo cual, unido a los hallazgos atípicos o con mínima o nula atrofia en las neuroimágenes, nos llevó a sospechar de otras etiologías y a proponer corticotomía y/o desconexiones localizadas en las 3 pacientes. La evolución hacia EPC, la progresión del déficit neurológico y de las neuroimágenes y/o hallazgos de actividad epileptiforme residual y/o más extensa en el video-EEG de superficie de control o en la ECoG intraoperatoria nos hizo sospechar que nos encontrábamos ante casos de ERIT. Esta sospecha se acentuó, al mantenerse restringida la afectación clínica y paraclínica en un solo hemisferio; incluso se ensayó inmunoterapia en 2 de las 3 pacientes antes de las reintervenciones quirúrgicas sin lograr una mejoría. En la tercera paciente tampoco se obtuvo una mejoría adicional con la inmunoterapia posterior al diagnóstico histopatológico confirmatorio. Los diagnósticos definitivos se establecieron en base los resultados típicos de la anatomopatología e inmunohistoquímica; como se establece en los criterios de Bien et al y Olson et al., 4,12.
Respecto al deterioro neurológico, una de nuestras pacientes desarrolló psicosis crónica; una entidad con mayor frecuencia en epilepsias farmacorresistentes que en epilepsia farmacosensible y también ha sido reportada la psicosis en la evolución de ERIT 14. Los 3 casos descritos cursaron con hemiparesia contralateral al hemisferio afectado. Sin embargo, en el caso 2, la paciente ya tenía hemiparesia derecha desde 6 años antes del inicio de las crisis epilépticas. Este dato podría considerarse como una etapa de pródromo de la evolución clásica de la ER de inicio infantil, en las cuales puede existir hemiparesia antes del desarrollo de epilepsia farmacorresistente o de la EPC; sin embargo, la hemiparesia de la paciente fue de inicio agudo y mejoró inicialmente con terapia física, sugiriendo a nuestro razonamiento, un origen por lesión vascular independiente de la ER. Posterior al desarrollo de la EPC su hemiparesia secuelar empeoró y se acentuó un poco más tras la cirugía. Respecto a ello, se ha descrito que lesiones cerebrales unilaterales adquiridas a temprana edad son más frecuentes en pacientes con ER comparados a pacientes sanos y que estas lesiones son ipsilaterales al hemisferio afectado por ER 15. De la misma forma, la paciente 3 presentó 2 lesiones identificadas en las neuroimágenes, sin que estas patologías pudieran explicar su cuadro clínico; pues los angiomas venosos no suelen asociarse al desarrollo de epilepsia y las calcificaciones residuales no se asocian a epilepsia farmacorresistentes; en ambos casos podrían ser silentes.
Finalmente, en los 3 casos, se ha evidenciado respuesta a la cirugía de epilepsia; evidenciándose la mejor respuesta a la hemisferotomía funcional en el caso 1, que las resecciones y/o desconexiones parciales (casos 2 y 3). En los casos 2 y 3 se ha desestimado ir a una hemisferotomía funcional por ser ambas pacientes diestras y el hemisferio afectado es el izquierdo, además de considerar la edad de la paciente 3, que condiciona a una menor posibilidad de recuperación de algún déficit cortical postquirúrgico. En nuestros 3 casos, como ya fue mencionado, no hubo respuesta a inmunoterapia. En la mayoría de los casos de niños y adultos, se han descrito respuestas pobres o solo transitorias a las inmunoterapias (corticoides, IgEV y recambio plasmático) tanto en la estabilización de los déficits neurológicos como en el control de crisis, en tanto que el éxito en el control de crisis mediante hemisferectomía o hemisferotomía funcional ha sido reportada hasta en un 80% (1-3,5,7, 10,11,13,16). En años recientes, se ha ensayado inmunoterapias en combinación y más agresivas (ácido micofenólico, tacrolimus, rituximab, alemtuzumab, adalimumab) en pequeñas series de pacientes o reportes de casos, con mejores resultados, pero con más efectos adversos o pobre perfil de seguridad en algunos de ellas (tacrolimus y alemtuzumab) y se desconoce la respuesta a largo plazo 1,2,5. También han sido reportados buenos resultados en resecciones más localizadas en algunos reportes de casos con pocos pacientes 3,18,19.
En conclusión, se debe tener en cuenta el diagnóstico de ERIT en pacientes adolescentes o adultos, cuyas epilepsias focales farmacorresistentes evolucionen a EPC, considerando que los déficits neurológicos focales, el deterioro cognitivo y los hallazgos típicos de las neuroimágenes podrían no estar presentes o ser de escasa representación. Seria de utilidad seguir la propuesta de Oslon respecto a la revisión de los criterios diagnósticos clásicos en casos de ER con presentación atípica, como ocurre en ERIT. La cirugía resectiva y/o desconexiones parciales podrían ser consideradas en casos especiales con crisis muy discapacitantes, cuando la hemisferectomía o hemisferectomía no se considere una opción en los adultos.

