











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCION
El Síndrome de MELAS (SM) es una enfermedad mitocondrial, neurodegenerativa progresiva, caracterizada por encefalomiopatía, acidosis láctica e ictus recurrentes que se asemejan a eventos isquémicos formando el acrónimo de MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, stroke like episodes); fue Pavlakis et al. 1 en 1984 quien realizó las primeras descripciones. El cuadro clínico se puede manifestar además de lo descrito, por epilepsia, talla baja, diabetes mellitus, oftalmoplejía, hipoacusia neurosensorial, cardiomiopatía e insuficiencia renal. En 1990, Godo et al. 2reportaron que 80% de casos de SM se debían a la variante A3243G (transición de adenina a guanina en el punto 3243) del gen MT-TL1 (mitochondrially encoded tRNA-Leu (UUA/G) 1,2).
En el 2007, en Sidney, se realizó un estudio poblacional en adultos que encontró una prevalencia de la variante A3243G-MT-TL1 de 236/100000 habitantes, cuyo hallazgo común fue la hipoacusia neurosensorial 3). En el mismo año, en Finlandia, en menores de 18 años, se estudió el fenotipo y la variante A3243G-MT-TL1, se encontró una prevalencia de 18.4/100000 habitantes 4). En Japón, en el 2011 se registró una prevalencia de 0.18/100000 habitantes 5). Recientemente, un artículo de revisión incluyó una búsqueda sistemática de casos entre 1990 al 2021, encontrando 51 casos de SM en Latinoamérica, y de ellos un sólo caso peruano 6).
Presentamos una familia con dos hermanos afectados con MELAS que presentaron estado epiléptico recurrente durante la evolución de la enfermedad.
Caso 1
Varón (Figura 1(A: III1) y (B)) que desde los 9 años, inició con fatiga ante los ejercicios habituales. Seis meses después presentó un estado epiléptico generalizado, seguido de crisis clónica del brazo derecho, que se repitió 8 meses después asociado a fiebre. Dos días después, presenta crisis tónico clónica del brazo derecho, seguido de agitación psicomotriz, disartria y amaurosis fugaz con recuperación en lapso de una semana. Dos meses después, presenta episodio que inició con cefalea seguido de nuevo estado epiléptico y hemiparesia izquierda que remitió en el lapso de un mes. Cinco meses después, presentó cuarto estado epiléptico, caracterizado por crisis clónicas del brazo izquierdo, con vómitos explosivos y disartria. Un mes después, sucedió el quinto estado epiléptico con crisis focales del brazo y hemicara izquierda acompañado de afasia de expresión que duró un mes en recuperarse. Y, tres meses después, ocurrió en sexto estado epiléptico, que fue semejante a los anteriores, con crisis focales en el brazo y hemicara izquierdos, esta vez, acompañado de conducta pueril y dislexia que persistió por 3 a 4 meses e hipoacusia persistente (tabla 1). Durante este tiempo se asoció valproato y clobazam al tratamiento con levetiracetam, en un control ambulatorio se retiró valproato. El resumen cronológico de síntomas y exámenes realizados se muestran en la tabla 1.
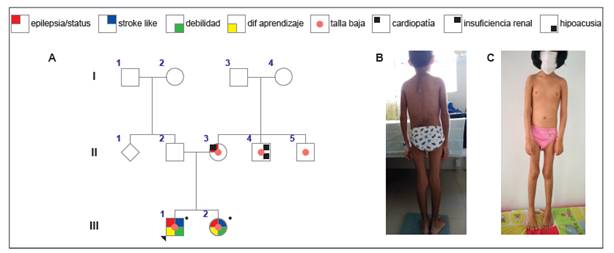
Figura 1 A. Heredograma. Caso 1: III-1, caso 2: III-2, (*) con estudio genético. B. Caso 1, niño de 10 años, se aprecia hipotrofia muscular e hirsutismo. C. Caso 2, niña de 9 años se aprecia hipotrofia muscular e hirsutismo
Tabla 1 Cronología de las manifestaciones clínicas, tratamiento y neuroimágenes
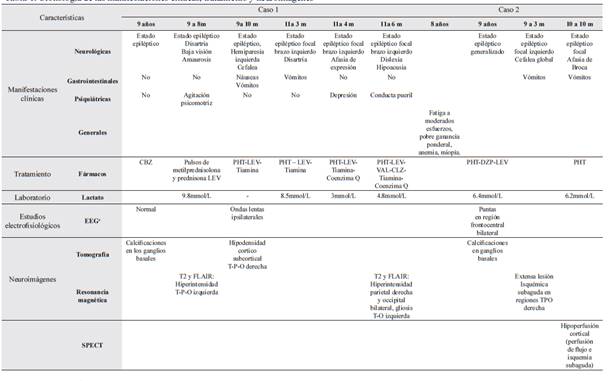
*Electroencefalograma b SPECT:Tomografía computarizada por emisión de fotón único Siglas: PHT: fenitoína, DZP: diazepam, LEV: levetiracetam, CLZ: clonazepam, VAL: valproato T: temporal, P: parietal, O: occipital.
Examen neurológico realizado a los 11 años 6 meses, evidencia funciones vitales estables, el índice de masa corporal (IMC) 12 (< 3DS), talla 115cm. (<3 DS), perímetro cefálico 47.5cm (<p3). Adelgazado, alerta, orientado, memoria conservada, lenguaje adecuado para la edad; podía hacer operaciones aritméticas correctamente hasta la multiplicación, hirsutismo, cuadriparesia 4/5 a predominio proximal, con hipotrofia muscular e hipotonía generalizada, normorreflexia osteotendinosa, baja visión e hipoacusia, reflejos cutáneos superficiales normales, sensibilidad táctil y dolorosa conservada, sin dismetría y marcha normal.
Los resultados de los exámenes de laboratorio fueron anodinos en todos los episodios de estado epiléptico; excepto, los niveles de lactato, los cuales fluctuaron entre 3 a 9,8mmol/L. La tomografía cerebral (TC) mostró calcificaciones bilaterales de núcleos de la base. Estudio de LCR realizado luego de segundo estado epiléptico fue normal. La Resonancia magnética cerebral (RMC) mostró hiperintensidad en el área temporo parieto occipital izquierdo en las secuencias T2 y FLAIR. La TC durante tercer estado epiléptico mostró lesión informada como infarto cerebral temporooccipital derecho. El electroencefalograma (EEG) inicial fue normal, y un segundo, durante el tercer estado epiléptico presentó ondas lentas en hemisferio derecho. La secuenciación NGS “next generation sequencing” de ADN mitocondrial identificó la variante m.3243A> G en el gen MT-TL1 con 66,2% de heteroplasmia compatible con SM.
Último control a los 13 años, con leve mejoría de la disartria, niega nuevas crisis epilépticas; recibe tratamiento con coenzima Q, tiamina, levetiracetam, clonazepam; y desde hace 1 mes recibe Arginina. Tiene discapacidad intelectual leve, y acude a colegio como alumno inclusivo.
Caso 2
Niña que desde los 8 años (Figura 1 (A: III2) y (C)) presenta fatiga a moderados esfuerzos, pobre ganancia ponderal y anemia. A los 9 años tuvo un estado epiléptico generalizado asociado a vómitos biliosos, fue tratada con fenitoína y luego con levetiracetam. Tres meses después, presentó un nuevo estado epiléptico clónico del brazo izquierdo, asociado a cefalea y vómitos, recibió fenitoína. Un año y medio después, ocurrió el tercer estado epiléptico focal con cefalea, vómitos, afasia, e hiperlactacidemia. El resumen cronológico de síntomas y exámenes realizados se muestran en la tabla 1.
El examen físico realizado a los 10 años 11 meses, evidenció peso 11.7 kg (<3DS), talla 114cm (<3DS), IMC 9 (< 3 DS) y perímetro cefálico 47cm (<p3). Adelgazada y de talla baja, alerta, con hirsutismo, monoparesia 4/5 en el brazo derecho, hipotrofia muscular generalizada, reflejos osteotendinosos – bicipital derecho, baja visión, hipoacusia bilateral y marcha normal.
Exámenes auxiliares: Hematimetría y bioquímica con hiperlactacidemia. La tomografía cerebral mostró calcificaciones en ganglios basales. El EEG fue anormal por actividad ictal epileptiforme. La RMN cerebral realizada a los 9 años, muestra una lesión tipo isquémica subaguda temporo parietooccipital derecha (figura 2). Estudio genético confirmó la presencia de la variante m.3243A> G en el gen MT-TL l con un 62% de heteroplasmia.
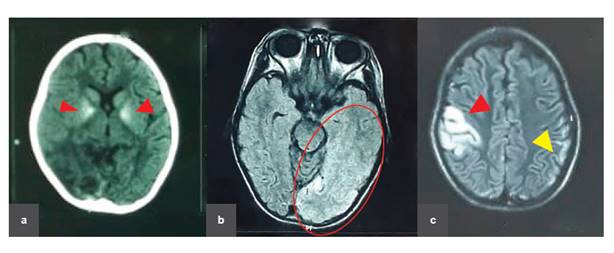
Figura 2 Neuroimágenes Caso 1 Tomografía cerebral, corte axial sin contraste. a) Tomografía cerebral (9años): Calcificaciones bilaterales en núcleos de la base. b) RMN cerebral, axial FLAIR (10años): hiperintensidad en región temporo parieto occipital izquierdo. c)RMN cerebral, axial FLAIR (10 años): hiperintensidad en región parieto temporal derecha e hiperintensidad e región parietal izquierda.
Último control realizado a los 11 años 10 meses con evolución estacionaria, sin crisis epilépticas, rendimiento académico bajo. Recibe tratamiento con tiamina, coenzima Q, levetiracetam, clonazepam y arginina.
DISCUSIÓN
Las variantes patogénicas en el ADN mitocondrial, como las que ocurren en el SM, ocasionan la producción insuficiente de energía para el correcto funcionamiento celular, generando múltiples enfermedades en los órganos de mayor demanda energética como el sistema nervioso, músculo esquelético, cardiaco, riñones, hígado y endocrino 7). El examen de oro para el diagnóstico de SM se realiza por secuenciación genómica mitocondrial 8). El origen de las variantes es heredada en el 80% de casos y de novo en el 20% a 25% restantes 9). La heteroplasmia se refiere a las diversas manifestaciones clínicas que un mismo individuo puede presentar de acuerdo al porcentaje de ADN mitocondrial mutado 10). En el caso 1 y 2 tienen heteroplasmia del 66.2%, y 62%, respectivamente.
La patogénesis del SM aún no está claramente definida; sin embargo, hay algunas teorías que intentan explicarlas. La teoría citopática mitocondrial propone una disfunción metabólica mitocondrial de las células de la musculatura lisa y las células endoteliales de las arterias cerebrales que pueden resultar en alteraciones en el calibre de los vasos, llevando a una angiopatía mitocondrial. La teoría de hiperexcitabilidad neuronal asume la disfunción del astrocito por deficiencia de ATP, causando alteración en la bomba Na+/K+ ATPasa y recaptación de glutamina; el excedente de potasio y glutamina en el espacio sináptico produce vasodilatación de los pequeños vasos cerebrales, produciendo muerte neuronal debido a excitotoxicidad 11).
La enfermedad tiene forma bimodal, la forma juvenil se presenta en promedio a los 9 años y la forma adulta a los 32 años 5). Los niños con SM tienen un neurodesarrollo temprano normal hasta el inicio de los síntomas, generalmente entre 2 a 10 años, como nuestros pacientes. Los primeros síntomas son la debilidad muscular en un 87%, fatigabilidad en un 15% a 18%, cefaleas recurrentes y crisis epilépticas en un 28% 12). La talla baja y la falla de medro son prevalentes en la forma juvenil. La hipoacusia, diabetes mellitus y hemiplejía se observan más en la forma adulta que en la juvenil 5), puede asociarse a mioclonus, atrofia óptica, falla cardiaca y oftalmoplejía externa progresiva 12). El tiempo de supervivencia es más corto en la forma juvenil que en la adulta, con un promedio de 20 años desde el inicio de la enfermedad 5); y en otros reportes el 29% de los pacientes que debutaron antes de los 20 años, tuvieron sólo 10 años de supervivencia 13).
Los pacientes con SM presentan disturbios en las funciones superiores, tales como inteligencia, lenguaje, percepción, atención y memoria; debido a los cambios metabólicos y/o degenerativos que se producen a lo largo del tiempo 7,14). Lo que se observó en el primer caso fue la pérdida de algunas habilidades adquiridas, y pasó de tener buen rendimiento escolar a presentar dislexia a los 11 años; así como agresividad, irritabilidad y conducta pueril. Estos cambios de comportamiento, nos orientan a pensar que se esté produciendo alteración de las funciones superiores, con posible deterioro cognitivo a largo plazo.
El 90% de casos de SM presentan alteración del estado mental y estados epilépticos. Las neuroimágenes muestran lesiones “stroke-like” que no respetan los territorios vasculares 7 y pueden acompañarse de afasia reversible, pérdida visual cortical y paresia, tal como se observó en ambos casos. Las lesiones cerebrales que se presentan en SM, se pueden encontrar en tres áreas que tienen en común una alta demanda metabólica: 1) la corteza visual pericalcarina (produciendo ceguera cortical o hemianopsia); 2) la corteza primaria somatosensorial (tercio medio del giro post central - región de mano y brazo) y 3) la corteza sensorial (vulnerable a injurias hipóxico isquémicas) 15). Los hallazgos de RMC en la etapa aguda son: hiposeñal en T1, hiperseñal en T2 y FLAIR (recuperación de inversión atenuada por fluído), patrón mixto en mapa ADC por edema vasogénico y citotóxico. En etapas sub agudas: hiperseñal en T1, hiposeñal en T2, mixto en FLAIR y patrón normal en difusión y ADC 16). Las lesiones que asemejan a infartos cerebrales se producen en áreas de alta demanda metabólica y no respetan los territorios vasculares. El edema citotóxico puede sobreponerse al edema vasogénico; la tumefacción edematosa puede ser reversible o puede evolucionar a encefalomalacia, sugiriendo un daño irreversible 17). El uso de la espectroscopía para la detección de ácido láctico en la zona lesional es de gran ayuda diagnóstica para MELAS 8). El SPECT nos permite detectar zonas de hipoperfusión en fases más tempranas que TC. En el caso 2, se realizó SPECT, determinando las áreas cerebrales comprometidas (figura 3).
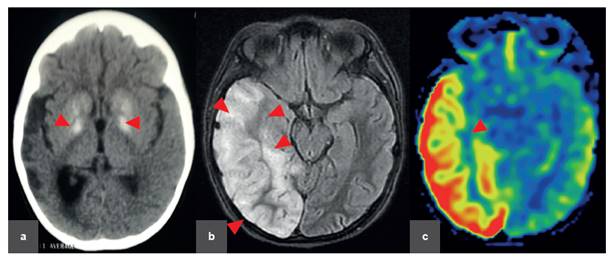
Figura 3 Neuroimágnes caso 2: a) Tomografía cerebral sin contraste (9a 11m): con calcificaciones bilaterales en núcleos de la base y cabeza de núcleo caudado bilateral. b) RMN cerebral, axial FLAIR: hiperintensidad en región temporo parieto occipital derecha. c) SPECT (10a 3m) Hipoperfusión en región temporo parieto occipital derecha.
Respecto a las opciones terapéuticas, no hay consenso de manejo, pero una serie de recomendaciones en base a casos publicados, indica manejo sintomático y terapia de soporte; tales como antiepilépticos, antioxidantes, suplementación de coenzimas para mejorar la función mitocondrial 8), implante coclear para la pérdida auditiva, analgésicos para la migraña, modificaciones en la dieta para el manejo de diabetes, ejercicio regular 7); y debe ser manejado por un equipo multidisciplinario (neurólogo, cardiólogo, endocrinólogo, genetista, audiólogo, oftalmólogo, terapista físico, psicólogo) 7). En el caso 1, durante un estado epiléptico, recibió ácido valproico; este fármaco puede alterar la membrana mitocondrial, induciendo cambios de despolarización paroxística, así como la alteración en la actividad de bombeo de protones del complejo IV (citocromo C oxidasa) en la cadena respiratoria; estos cambios pueden precipitar aún más la sintomatología 18), por lo que se decidió suspender.
Algunos reportes sugieren que al momento en que el paciente presenta síntomas sugestivos de infarto, se sugiere el uso de arginina intravenosa (0,5gr/kg), administrada en las primeras 3 horas del evento, asociado a bolos de cloruro de sodio al medio normal para mantener la perfusión cerebral; además de dextrosa, para detener el catabolismo (incluso si se encuentra euglicémico al momento de la evaluación); si el paciente persiste con trastorno del sensorio, lo indicado es realizar pronto un EEG, porque puede estar en un estado no convulsivo). Para el manejo de mantenimiento, se sugiere la administración de una dosis adicional de arginina EV 0.5gr/kg para infundirlo en 24 horas por los siguientes 3 a 5 días, luego que se estabilice puede seguir tomando arginina oral una vez al día 16). Como dosis profiláctica se recomienda arginina diaria 0,15-0,30g/kg repartido en 3 dosis 16).
El uso del “cocktails” vitamínicos y antioxidantes para enfermedades con déficit de energía es controversial, algunos artículos mencionan la utilidad del uso de la coenzima Q10 (ubiquinona), asociado a tiamina y riboflavina, como parte de la interacción en la cadena respiratoria 19), mientras otras referencias no sugieren su uso en SM 20). Una vez realizado el diagnóstico, deben hacerse controles regulares para evaluar el progreso y deterioro de la enfermedad; entre ellos, el examen neurológico detallado con evaluación neuropsicológica, resonancia cerebral, examen visual y auditivo, evaluación de crecimiento, descarte de hipotiroidismo y diabetes.
En conclusión, presentamos dos hermanos con SM de presentación juvenil asociada a la variante m.3243A > G-MT-TL1, con enfermedad caracterizada por estados epilépticos recurrentes, lactacidemia, déficit neurológico, lesiones semejantes a infartos cerebrales, falla de medro, microcefalia y pérdida de algunas habilidades adquiridas. Esta sintomatología debe considerarse en niños que presentan estado epiléptico, lactacidemia y lesiones que simulan infartos cerebrales.

