











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCION
La ataxia de Friedreich (AF) es una enfermedad neurodegenerativa poco conocida por los profesionales de salud neurólogos y no neurólogos. Esta revisión provee información actualizada y relevante sobre aspectos fisiopatológicos, clínicos y terapéuticos, para promover un diagnóstico oportuno y manejo adecuado en el personal sanitario. Se recopila información sobre la epidemiología incluyendo búsqueda sistemática de casos de AF en Latinoamérica, las manifestaciones clínicas, el diagnóstico y el tratamiento.
Definición y aspectos históricos
La AF es una ataxia hereditaria multisistémica de herencia autosómica recesiva caracterizada por una progresión de ataxia cerebelosa, disartria, piramidalismo, pérdida de sensibilidad profunda, cardiomiopatía hipertrófica, anormalidad musculoesqueléticas y diabetes1. La AF es causada por una expansión anormal de repeticiones GAA en el gen FXN ocasionando una disminución de la expresión de la proteína mitocondrial frataxina 2.
La AF fue descrita por primera vez por el médico alemán Nicholaus Friedreich en una serie de 5 artículos, reportes de 9 casos correspondientes a 3 familias entre los años 1863 a 1877 3,4. Friedreich quien fue profesor de Medicina en Heidelberg denominó a esta enfermedad como “atrofia de las columnas posteriores de la médula espinal” para referirse a una ataxia con edad de inicio alrededor de la pubertad, junto con escoliosis, deformidades en los pies, afectación cardiaca, y compromiso motor y sensitivo. Fue el neurólogo francés Pierre Marie quien décadas después, diferenció esta enfermedad de las ataxias dominantes y difundió los hallazgos de Friedreich en el mundo científico 5. En 1988, Chamberlain y colaboradores lograron localizar el locus asociado a AF en una región del cromosoma 9 6.
Epidemiología
La AF es considerada la ataxia hereditaria más frecuente a nivel mundial, con una prevalencia estimada en poblaciones caucásicas de 1:20 000 - 1:50 000 7. En algunas regiones se han descrito prevalencias muy bajas de AF como 1:750 000 en Finlandia y 1:330 000 en Rusia7,8. En las poblaciones africanas y asiáticas se considera una enfermedad muy rara y la prevalencia es mucho menor9.
Ataxia de Friedreich en Latinoamérica
Para determinar el impacto de la AF en Latinoamérica, realizamos una búsqueda sistemática de casos de Ataxia de Friedreich siguiendo las pautas PRISMA (Figura 1). Se realizó una búsqueda de estudios publicados en las bases de datos de MEDLINE, LILACS y SciELO. La búsqueda incluyó los siguientes términos: “Friedreich Familiar Ataxia”, “Friedreich Spinocerebellar Ataxia”, “Friedreich's Ataxia”, “Friedreich Disease”, “GAA expansión”, “Hereditary Spinal Sclerosis”, restringiéndose la búsqueda para Latinoamérica y el Caribe con los siguientes términos "Hispanic or Latino", "Latin American" “Hispanic Americans”, así como búsqueda por cada país de la región. La búsqueda fue similar con términos en español para las bases de datos regionales. Para la búsqueda se aplicaron los siguientes criterios de inclusión: 1) estudios observacionales, series de caso o reportes de caso que incluyan pacientes con diagnóstico genético de ataxia de Friedreich sin restricciones de edad. 2) Casos de ataxia de Friedreich en población Latinoamericana o del Caribe. 3) Estudios que consideren datos sociodemográficos y clínicos, individuales o agregados. 4) Estudios publicados hasta el 28 de enero de 2023.
Se utilizó el software Rayyan para eliminar duplicados y someter al screening por título y abstract. Posteriormente se revisaron los estudios a texto completo y se seleccionaron aquellos que cumplieron con los criterios de selección. Los datos se extrajeron en una base de datos Excel. Los datos extraídos incluyeron: país, nombre del primer autor, año de publicación, número de casos / número de familias, edad de inicio de la enfermedad, sexo, cuadro clínico y diagnóstico genético y se resumen en la Tabla 1.
Tabla 1. Ataxia de Friedreich en Latinoamérica, búsqueda sistemática de casos.
| País Autor, año | N° casos / N° familias | Edad de inicio/ Sexo | Cuadro clínico | Diagnóstico genético |
|---|---|---|---|---|
| Brasil Albano et al, 2002(10) | 17 casos / 15 familias | 9,8 ± 4,1 años | ND | 17 casos con expansión homocigota |
| Brasil Zeigelboim et al, 2017(11) | 30 casos / ND | 10 F/ 20 M | ND | Todos con diagnóstico genético, homocigotas |
| Brasil Rezende et al, 2019(12) | 37 casos/ ND | 9.2 ± 2.6 años 26 F / 11 M | ND | Todos con diagnóstico genético, homocigotas |
| México Boll et al, 2021(13) | 23 casos / ND | 15.6 ± 3.8 años 12 F / 11 M | ND | 18 con expansión homocigota 5 con expansión heterocigota |
| Perú Cornejo-Olivas et al, 2020(14) | 9 casos / 9 familias | ND | ND | 7 con expansión homocigota 2 con expansión heterocigota |
| Brasil Peluzzo et al, 2019(15) | 141 casos / 128 familias | ND | ND | 138 homocigotos y 3 heterocigotos compuestos |
| Brasil Fussiger et al, 2018(16) | 27 casos / 27 familias | ND | Presencia de fenotipos: parcial, clásico, LOFA y FARR | 26 homocigotos y 1 heterocigoto compuesto |
| Brasil Rezende et al 2023(17) ENIGMA-Ataxia Working Group | 256 casos / ND | 17 ± 9 años 131 F / 114 M | ND | Todos los diagnósticos de fueron confirmados genéticamente |
| Brasil Harding et al, 2021(18) ENIGMA-Ataxia Working Group | 248 casos / ND | 16.7 ± 9.2 años 128 F / 120 M | ND | 243 con expansión homocigota 5 con expansión heterocigota compuesta |
| Brasil Takazaki et al, 2018(19) | 28 casos / ND | ND 17 F / 11 M | 19 casos con fenotipo clásico y 9 casos con fenotipo LOFA | 28 con expansión homocigota |
| Brasil Takazaki et al, 2021(20) | 27 casos / ND | ND 15 F / 12 M | 24 casos con fenotipo clásico y 3 con fenotipo LOFA | 27 casos con expansión homocigota |
| Brasil Da Silva et al, 2013(21) | 22 casos / ND | 15.0 ± 5.1 años 14 F / 8 M | 20 casos con fenotipo clásico, 1 con fenotipo FARR y 1 con fenotipo LOFA | 22 casos con expansión homocigota |
| Brasil Vieira et al, 2015(22) | 21 casos / ND | 13.8 ± 8.7 años 13 F / 8 M | Ataxia a la marcha, ataxia en extremidades, disartria, pérdida de sensibilidad, arreflexia, deformidad en pies, etc. | 21 casos con más de 100 repeticiones GAA homocigotas |
| Brasil Rezende et al, 2017(23) | 32 casos / ND | 12.0 ± 8.0 años 20 F / 16 M | 23 casos con fenotipo clásico, 13 con fenotipo LOFA | 36 casos con expansión homocigota |
| Brasil Rezende et al, 2015(24) | 31 casos / ND | 12.0 ± 8.0 años 20 F / 11 M | 29 casos con fenotipo clásico, 2 con fenotipo LOFA | 31 casos con expansión homocigota |
| Brasil Zeigelboim et al, 2018(25) | 30 casos / ND | ND 10 F/ 20 M | ND | 30 casos con diagnóstico genético |
| Brasil Bonilha da Silva et al, 2014(26) | 35 casos / ND | 13.3 ± 5.2 años 22 F/ 13 M | ND | 35 casos con expansión homocigota |
| Brasil Franca et al, 2009(27) | 24 casos / ND | 13 F/ 11 M | 18 casos con fenotipo clásico, 5 con fenotipo FARR y 1 con fenotipo LOFA | 24 casos con expansión homocigota |
| Brasil Musegante et al, 2013(28) | 158 casos / ND | ND | ND | Todos con diagnóstico genético |
| Brasil Albano et al, 2001(29) | 17 casos / 15 familias | 10 años ND | ND | 17 con expansión homocigota |
| Brasil Salomao et al, 2017(30) | 1 caso | 28 años M | Ataxia a la marcha, pie cavo bilateral, atrofia de músculos peroneos, ausencia de reflejo tendinoso profundo y pérdida propioceptiva. Escoliosis leve | Expansión FXN con 66 repeticiones GAA |
| Brasil Rangel et al, 2019(31) | 7 casos / ND | ND | ND | 5 casos con con expansión homocigota y 2 casos heterocigotos |
| Brasil Moro et al, 2014(32) | 3 casos / 1 familia | 36 años 1 F / 2 M | Ataxia a la marcha, disartria, ausencia de reflejos, disfagia, | 3 casos con diagnóstico genético |
| Argentina Pérez et al, 2013(33) | 2 casos / 1 familia | 33 años 1 F / 1 M | ataxia axial y apendicular, disartria, arreflexia | 2 con expansión homocigota |
| Brasil Cardozo et al, 2020(34) | 20 casos / ND | ND 11 F / 9 M | ND | 20 con diagnóstico genético |
| Brasil Schwartz et al, 1999(35) | 5 casos / ND | 14 años 3 F / 2 M | ataxia axial y apendicular, disartria, arreflexia | 5 con expansión homocigota |
| Brasil Martinez et al, 2017(36) | 106 casos / ND | 13.5 ± 5.0 años 61 F/ 45 M | 88 casos con fenotipo clásico, 18 con fenotipo LOFA | 106 casos con diagnóstico genético |
| Colombia Pedraza et al, 2013(37) | 24 casos/ ND | ND | ND | 24 con diagnóstico genético |
| México Cervantes et al, 2009(38) | 1 caso | 15 años F | ND | 1 con expansión homocigota |
| Brasil Abrahao et al, 2016(39) | 11 casos / 10 familias | 8-20 años 9 F / 2 M | ND | 11 con expansión homocigota |
| Brasil Santos et al, 2015(40) | 20 casos / ND | 15,4 ± 5,2 años 12 F / 8 M | ND | 20 con diagnóstico genético |
| México Rasmussen et al, 2006(41) | 14 casos / ND | ND | ND | 14 con expansión homocigota |
| México Gómez et al, 2004(42) | 14 casos / 12 familias | ND | ND | 14 con expansión homocigota |
| Cuba Cruz-Mariño et al, 2010(43) | 3 casos / 2 familias | 11 años 2F / 1M | Debilidad progresiva, ataxia axial y apendicular, disartria, arreflexia | 3 con diagnóstico genético |
| Brasil Chevis et al, 2012(44) | 33 casos / | ND 18 F / 15 M | ND | 33 con expansión homocigota |
ND: no disponible, LOFA: late onset Friedreich’s ataxia, FARR: Friedreich’s ataxia with retained reflexes
El resultado de la búsqueda en las bases consideradas dio como resultado 193 estudios, 53 de ellos fueron evaluados a texto completo, quedando únicamente 35 estudios (10-44) que cumplieron con los criterios de selección. La selección de estudios se muestra en el diagrama de flujo PRISMA correspondiente.
Los 35 estudios corresponden a publicaciones en Brasil (1391 casos), México (52 casos), Colombia (24 casos), Perú (9 casos), Argentina (2 casos) y Cuba (3 casos), tal como se detalla en la figura 2. Es importante señalar que varios de los estudios publicados en Brasil corresponden a poblaciones temporal y geográficamente similares, por lo cual es probable que el número total de casos para dicho país esté sobreestimado. El total de casos con diagnóstico genético fue de 1481, de los cuales 1463 corresponden a casos homocigotas y 18 a casos heterocigotas compuestos. Respecto al sexo, existe una ligera predominancia en el sexo femenino, alcanzando un 54.6 % de los casos reportados para Latinoamérica. La edad de inicio se encontró que es de 15,2 ± 7,7 años, sin embargo, esta estimación no diferencia los fenotipos clásicos de los de inicio tardío, lo cual se debe tener en cuenta al momento de hacer comparaciones con poblaciones específicas.
GENÉTICA
La AF es una enfermedad autosómica recesiva causada por la expansión anormal de repeticiones GAA ubicadas en el primer intrón del gen FXN (Cr9q21.11), que codifica una proteína denominada frataxina45. Alrededor del 94% de individuos afectados tiene una expansión bialélica anormal; mientras que el resto presenta una expansión anormal en un alelo y una variante patogénica diferente en el otro 46.
Los alelos FXN en la población general contienen entre 5 a ~30 repeticiones GAA; mientras que los pacientes AF presentan alelos de ~70 repeticiones GAA a más. No obstante, los límites del rango en la clasificación de alelos no están totalmente definidos debido a fenotipos intermedios y tamaño de alelos fuera del rango propuesto47.
Los mecanismos que conllevan a la degeneración en AF están relacionados fundamentalmente a la pérdida de función a causa de un silenciamiento génico a nivel transcripcional48. La expansión anormal de GAA genera cambios estructurales como la formación de loops y la heterocromatización anormal debido a la metilación del ADN y a la modificación de histonas por desacetilación o metilación 49. Estos cambios afectan el funcionamiento de la ARN polimerasa II, conllevan a una reducción del total de ARNm transcritos y menor traducción de la proteína frataxina.50 A diferencia de otras enfermedades por expansión de microsatélites, la AF no comparte la característica conocida como anticipación genética, debido a su naturaleza autosómica recesiva 46.
FISIOPATOLOGÍA
La frataxina (FXN) es una proteína compuesta por 210 aminoácidos, que actúa a nivel de la matriz mitocondrial 51. La FXN se distribuye predominantemente en tejidos con alto gasto metabólico como el corazón, el cerebro, el hígado, el páncreas y el músculo esquelético 52. Se han asignado varias funciones a la FXN, demostrando que estaría involucrada principalmente en el metabolismo celular y la homeostasis del hierro 53.
La FXN participa en la activación del ensamblaje de los clusters hierro- sulfuro o ISC, que están compuestos de 2 o 4 átomos de hierro enlazados con 2 o 4 átomos de sulfuro [2Fe-2S] [4Fe-4S] 54. El ensamblaje de hierro y sulfuro ocurre en un complejo proteico, conformado por la proteína ISCU (Iron-sulfur cluster assembly) que actúa como andamio, la proteína transportadora de acilo (ACP) y una cisteína desulfurada (NFS1) que dona átomos de sulfuro con la ayuda de una proteína estabilizadora ISD 11. La FXN promovería la transferencia de azufre de la NFS1 a la proteína de andamiaje ISCU, así como la donación de átomos de Fe 55. Los grupos ISC participan en el transporte de electrones en los complejos I y II de la cadena respiratoria, donde gracias a un proceso de fosforilación oxidativa genera ATP 52.
La deficiencia de la FXN que ocurre en la AF disminuye la formación de grupos ISC. Esto se manifiesta en la disminución de la producción de adenosina trifosfato (ATP) y una acumulación excesiva de hierro mitocondrial 56. El hierro libre a través de la reacción química de Fenton activa la producción de especies reactivas de oxígeno (ROS) en las mitocondrias, llevando a la muerte celular 57. Estos mecanismos fisiopatológicos que conllevan a la neurodegeneración en AF se muestran en la Figura 3.
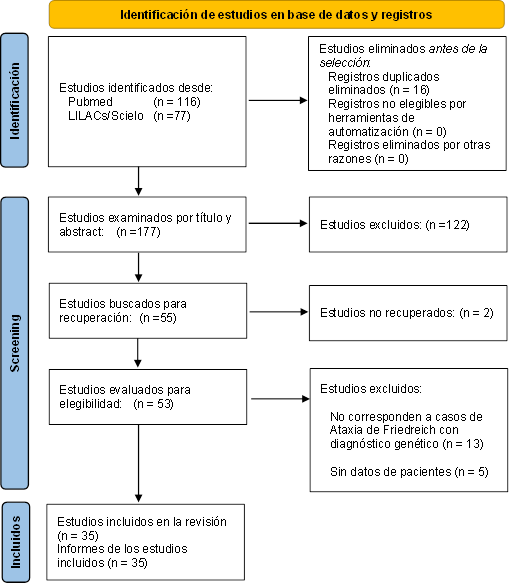
Figura 1 Diagrama de flujo para la búsqueda sistemática de casos de Ataxia de Friedreich en Latinoamérica
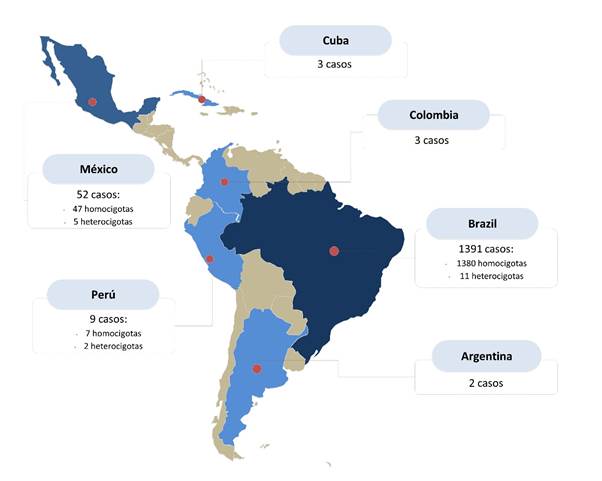
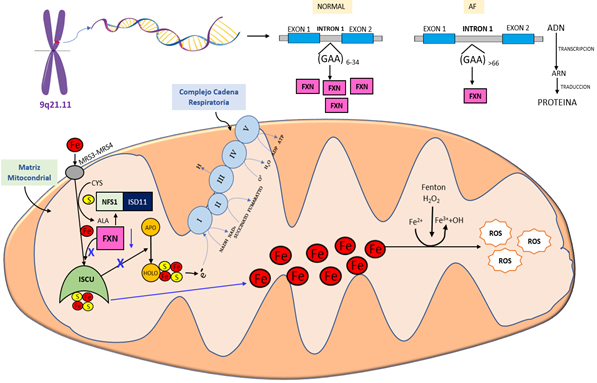
Figura 3 Neurodegeneración en Ataxia de Friedreich, principales mecanismos. FXN: Proteína Frataxina, Fe: Hierro, S: Sulfuro, ISCU: Proteína Iron-sulfur cluster assembly , NFS1: cisteína desulfurada , ISD 11: proteína chaperona ISD11, Cys: Cisteina, Ala: Alanina, ROS: reactive oxygen species.
La neuroinflamación sería otro mecanismo de neurodegeneración en AF 58. Ante el daño oxidativo y la ferroptosis se produce un aumento en la activación de la microglía y los astrocitos; células de defensa del sistema nervioso que producen citocinas, quimiocinas y especies reactivas de oxígeno 59. En la AF la neuro inflamación crónica altera el equilibrio entre la formación de especies reactivas y la defensa antioxidante endógena, incrementando la muerte neuronal. En la AF también se compromete la remielinización, siendo la oligodendroglia y las células de Schwann las más susceptibles al déficit de FXN 60.
La neurodegeneración ocurre tanto en el sistema nervioso central como en el periférico, afectando principalmente los ganglios de la raíz dorsal, las columnas posteriores, provocando desmielinización de los tractos espinocerebelosos y atrofia de los núcleos dentados, esto debido a que en el sistema nervioso la expresión de la FXN es mayor en los ganglios de la raíz dorsal y en la capa granular del cerebelo 61.
MANIFESTACIONES CLÍNICAS
La AF es una enfermedad multisistémica que afecta principalmente el sistema nervioso, tanto central como periférico; así como los sistemas musculoesquelético, cardiovascular y endocrino. En una cohorte europea se encontró que los síntomas iniciales neurológicos correspondieron a un 90,7% y los síntomas no neurológicos en un 9,3% de los casos 62.
Manifestaciones neurológicas
La ataxia es el síndrome cardinal en AF. La ataxia es resultado de una disfunción combinada del cerebelo, los cordones posteriores y los tractos espinocerebelosos de la médula espinal, los nervios periféricos y en algunas ocasiones, el sistema vestibular. Se presenta precozmente en todos los casos de AF con una progresión lenta. Se evidencia a nivel apendicular, con dismetría y disdiadococinesia, y a nivel axial, con inestabilidad durante la bipedestación y la marcha(50). La disartria cerebelosa, con disfunción muscular laríngea o velofaríngea, se presenta en un 70% de casos de AF con lenta progresión33,63.
Las alteraciones sensitivas se producen debido al compromiso de la médula espinal, la radiculopatía y la neuropatía periférica. La degeneración temprana de la columna posterior de la médula espinal compromete la propiocepción, produciendo predominantemente hipopalestesia, abatiestesia y dificultad en la discriminación entre dos puntos 64.
La arreflexia es un síntoma predominante en AF. Los reflejos osteotendinosos se encuentran disminuidos o abolidos en alrededor de un 90% de pacientes con AF. Esta afectación ocurre por la degeneración precoz de los ganglios de la raíz dorsal y la neuropatía periférica. Los pacientes con fenotipos de inicio tardío suelen preservar los reflejos osteotendinosos 65.
Los signos piramidales se producen por afectación de la vía corticoespinal a nivel de la médula espinal. Las respuestas plantares extensoras están presentes hasta en 70 a 90% de los casos de AF. No obstante, la espasticidad y la hiperreflexia no suelen evidenciarse clínicamente, ya que están enmascaradas por el compromiso de la segunda motoneurona. En las formas tardías de AF se puede apreciar una mayor frecuencia de espasticidad, con contracturas y espasmos musculares. En estadios avanzados de la enfermedad puede observarse paraparesia, o incluso cuadriparesia 66.
Se han descrito alteraciones de la vía sensorial visual en la AF. Si bien la mayoría de los pacientes con AF no mencionan síntomas visuales, hasta un 30% presenta signos de disfunción visual durante la evaluación clínica como: una menor sensibilidad al contraste, disminución de la agudeza visual y defectos campimétricos 66. Se ha documentado que hasta 90% de casos AF presentan alguna anormalidad documentada en los potenciales evocados visuales 67. Las imágenes por tomografía de coherencia óptica muestran una reducción de la capa de fibras nerviosas, con disminución del grosor en todos los cuadrantes, con respeto de la mácula y regiones circundantes. En formas más severas de AF, asociadas a grandes expansiones de repeticiones, hay una evolución más rápida del compromiso visual semejante a la neuropatía óptica hereditaria de Leber 68.
Las alteraciones del sistema oculomotor son comunes en la AF. La mayoría de las anomalías oculomotoras se explican por el compromiso del tronco encefálico, el cerebelo y sus conexiones; y se producen en alrededor del 40% de los pacientes 66,69. En AF la fijación ocular se interrumpe con intrusiones sacádicas, como las sacadas de onda cuadrada y flutter ocular. Las sacadas de onda cuadrada usualmente horizontales se producen tanto en la mirada primaria, durante la fijación y en el seguimiento lento. Estas pueden tener, con menos frecuencia, un componente vertical asociado y ser oblicuas, o presentarse verticales de manera aislada. El nistagmo provocado por la mirada horizontal lateral es el más frecuentemente encontrado en AF y se produce en alrededor del 60% de los casos 69. Otras alteraciones oculomotoras son las que se producen durante los movimientos sacádicos como la latencia prolongada y la dismetría 70. En un estudio de 20 pacientes con AF, se observó que en promedio el 54% de las sacadas fueron adecuadas, 37% hipermétricas y 9% hipométricas 71.
La AF asocia alteraciones neuro-otológicas como disfunción vestibular e hipoacusia. La latencia prolongada de los reflejos vestíbulo-oculares son características en AF y se produce por el compromiso del núcleo vestibular del tronco encefálico y del VIII nervio craneal 72. Hasta un 25% de casos de AF presentan oscilopsia en relación a afectación vestibular bilateral 71. Alrededor del 8-10% de pacientes con AF cursa con pérdida progresiva de la audición con diversos grados de alteración en potenciales evocados auditivos 66,73.
Las manifestaciones autonómicas en AF están relacionadas principalmente con la micción y la defecación. Se presentan tanto retención como urgencia urinarias, resultado de la hipoactividad o hiperactividad del músculo detrusor de la vejiga 28. Los pacientes con AF frecuentemente presentan estreñimiento, relacionado con una motilidad intestinal reducida, o incontinencia fecal. Otras manifestaciones disautonómicas son la bradicardia, la dishidrosis y cambios en la piel por alteraciones vasomotoras con frialdad distal y cianosis 50.
La AF compromete también las funciones cognitivas en grado variable. El compromiso disejecutivo incluye alteraciones en la velocidad de procesamiento de información, memoria de trabajo, abstracción, atención y razonamiento. El compromiso cognitivo se ha relacionado con una afectación en las conexiones entre el cerebelo y las áreas prefrontales, así como de redes más extensas que involucraría la protuberancia, el tálamo y otras áreas corticales. Dentro de las áreas corticales que se verían afectadas sería la corteza parietal, lo que explicaría las alteraciones en la capacidad visoconstructiva y visuoespacial de los pacientes 48,50.
Los pacientes con AF experimentan síntomas psiquiátricos diversos. La depresión es tanto reactiva a la enfermedad como secundaría a la neurodegeneración de varios núcleos encefálicos. Las formas severas de depresión se presentan hasta en un 10% de los pacientes con AF 50. Se han descrito otras manifestaciones clínicas diversas incluyendo alteraciones del sueño, coexistencia de cefaleas primarias como migraña, crisis epilépticas entre otros 66.
Manifestaciones sistémicas
Las manifestaciones sistémicas incluyen grados variables de cardiomiopatía, los trastornos de la conducción, diabetes mellitus, la escoliosis y las deformidades musculoesqueléticas.
El compromiso cardíaco es frecuente y repercute considerablemente en la morbimortalidad de los pacientes con AF. La cardiopatía es la primera causa de mortalidad en AF y es más frecuente y severo en formas tempranas con mayores expansiones GAA 74. La miocardiopatía se presenta en hasta el 40% de los casos, posiblemente asociada a fenómenos de miocarditis con acumulación de hierro y estrés oxidativo que causan muerte celular, pérdida de las proteínas contráctiles y una remodelación cardiaca por fibrosis 66,75. La cardiomiopatía hipertrófica concéntrica es la forma más común de cardiomiopatía, con mayor afección del ventrículo izquierdo. Se ha observado que pacientes AF que iniciaron con cardiomiopatía hipertrófica luego devinieron en una cardiomiopatía dilatada asociada a mayor mortalidad 76. La fracción de eyección puede tener una evolución tórpida hasta en 22% de los pacientes 77. La remodelación cardiaca favorece a la aparición de trastornos de la conducción. Los pacientes AF suelen presentar alteraciones de la repolarización en derivadas inferiores y laterales del ECG hasta en un 85% de casos. Se han registrado bloqueos de rama en aproximadamente 18% de casos 77. Las arritmias auriculares como el flutter y la fibrilación auricular se describen hasta en 5% de casos, con menor ocurrencia de arritmias ventriculares 66. Al inicio de la enfermedad miocárdica, el paciente suele mantenerse asintomático, con una adecuada fracción de eyección; sin embargo, puede disminuir considerablemente poco antes de su muerte 78.
La diabetes es el trastorno metabólico más reportado en pacientes con AF. Se presenta en alrededor del 8%, con mayor frecuencia y formas más severas en aquellos que iniciaron con síntomas de AF antes de los 25 años 66. El diagnóstico de diabetes en AF suele realizarse en la edad adulta 79. La aparición de diabetes en AF se explica por la disfunción mitocondrial desencadenada por la deficiencia de frataxina, que alteran las señales de estímulo para la secreción de insulina. Entre otras explicaciones se encuentran el estrés oxidativo, y la activación de señales proapoptóticas que destruyen las células B pancreáticas. La disfunción mitocondrial en el hígado, los adipocitos y en el músculo esquelético conlleva a un incremento de la adiposidad corporal y a la resistencia a la insulina 80. Otras alteraciones endocrinas en AF incluyen hipotiroidismo en aproximadamente 4% de los paciente, particularmente en la AF de inicio tardío 66.
La escoliosis y las deformaciones de los pies son las manifestaciones musculoesqueléticas más características en los pacientes con AF. La localización más común de la escoliosis es la torácica, seguida de la lumbar. Hasta un 90% de los pacientes menores de 15 años presentan escoliosis moderada a severa, que incluso pueden requerir de cirugía 81. Se ha descrito hipercifosis hasta en un 66% de casos. Las deformidades en los pies se presentan hasta en el 57% de casos de AF 66.
FENOTIPOS CLÍNICOS EN AF
La AF típica está caracterizada por ataxia mixta de la marcha y las extremidades, disartria, pérdida de reflejos de las extremidades inferiores y compromiso sensitivo. A esta presentación se le conoce como fenotipo clásico y se presenta hasta en 75% de los casos. En el fenotipo clásico, la edad de presentación se da entre los 10 y los 16 años, con una progresión lenta y con frecuente compromiso cardiaco 82. En este fenotipo son comunes la ausencia de reflejo rotuliano y aquíleo asociado a la respuesta plantar extensora. La espasticidad de miembros inferiores es también frecuente y se asocia a deformidad en equinovaro 65,66.
Los casos que difieren del fenotipo clásico se conocen como fenotipos atípicos y representan hasta el 25% de casos 83. Los fenotipos atípicos más conocidos de AF incluyen 1) la AF de inicio tardío o LOFA, 2) la AF de inicio muy tardío o VLOFA y 3) la variante con conservación de reflejos FARR 84.
En el fenotipo LOFA (del inglés Late Onset Friedreich Ataxia), la edad de inicio de síntomas ocurre después de los 25 años. Este fenotipo se asocia con expansiones menores de alrededor de 500 repeticiones GAA (85). Se caracteriza por espasticidad de extremidades inferiores, retención de reflejos, con menor compromiso cardíaco y esfinteriano 66. Aquellos casos que inician síntomas después de los 40 años, se les conoce como el fenotipo VLOFA (del inglés Very late onset Friedreich ataxia) asociado a expansiones de alrededor de 300 repeticiones GAA 85.
El fenotipo AF con retención de reflejos FAAR (del inglés Friedreich Ataxia with retained reflexes) se presenta en aproximadamente el 12% de los pacientes con expansión bialélica para la expansión GAA (65). La edad de inicio es variable con rangos entre los 13 a 45 años 86. En el fenotipo FAAR, los reflejos osteotendinosos pueden estar incrementados o incluso asociar clonus. Estos se asocian a una menor incidencia de miocardiopatía y compromiso musculoesquelético 65,85. Además de los fenotipos atípicos ya descritos, existen reportes aislados que propones fenotipos alternativos como: 1) AF de Arcadian descrito en diez familias acadianas de Nueva Brunswick, Canadá, caracterizada por ataxia leve con inicio tardío y menor ocurrencia de miocardiopatìa y diabetes 87; 2) Heterocigotos con variantes puntuales del gen FXN, que presentan un fenotipo clínico más leve, con una progresión lenta pero una marcha espástica de inicio temprano, reflejos conservados, sin disartria y sin ataxia; 3) Heterocigotos con grandes deleciones en FXN, fenotipo caracterizado por una progresión rápida con manifestaciones neurológicas severas, que asocian otros movimientos anormales como corea y temblor de intención; 4) AF de inicio temprano, con inicio en menores de diez años, se han reportado casos incluso en menores de 5 años, caracterizada por rápida progresión, baja estatura, extremidades cortas y una alta ocurrencia de miocardiopatía y diabetes. y 5) Miocardiopatía aislada, asociada a aparición muy tardía de la ataxia. 66,82
Las características clínicas entre fenotipos típicos y atípicos se resumen en la (Tabla 2).
Tabla 2 Características clínicas de AF por fenotipos Típico vs atípicos
| Manifestaciones Clínicas | Típico | Atípico (tardío) |
|---|---|---|
| Frecuencia (%) | 83.1 | 16.6% |
| Edad (años) | 29 ± 11.9 | 50 ± 10.4 |
| Edad de inicio (años) | 12 ± 4.4 | 32.5 ± 7.4 |
| Sexo Hombres/Mujeres | 47.6/52.4 | 42.6/57.4 |
| Duración de la enfermedad (años) | 17 ± 11.9. | 14 ± 7.4 |
| Estadio de Invalidez | 83.1 | 16.6 |
| Genetica: Repeticiones GAA (%) | 78.8 | 16.3 |
| Escoliosis Leve/mod/sev (%) | 34.6/32/13.7 | 25.9/6.5/1.9 |
| Deformidades de los pies Leve/mod/sev (%) | 11.9/27.6/24.6 | 7.4/14.8/1.9 |
| Cardiomiopatia (%) | 46.1 | 11.1 |
| Movimentos anormales de los ojos (%) | 90.7 | 88.9 |
| Disminución de la agudeza visual (%) | 37.2 | 34.3 |
| Perdida de la audición | 12.4 | 3.7 |
| Diabetes tipo 1-2 (%) | 4.8/3 | 0/3.7 |
| Hipotiroidismo (%) | 3.3 | 7.4 |
| Disfagia Leve/mod/sev (%) | 43.7/23.9/1.9 | 44.4/18.5/0.9 |
| Sintomas sensitivos pies Leve/mod/sev(%) | 22.7/31.6/36.6 | 32.1/41.5/18.9 |
| Paresia (%) | 65.9 | 25.9 |
| Miembros sup. Proximal Leve/mod/sev(%) | 18.1/5.2/1.5 | 2.8/0/0 |
| Miembros sup. Distal Leve/mod/sev(%) | 22.6/18.1/5.6 | 13.9/0.9/0 |
| Miembros inf. Proximal Leve/mod/sev(%) | 22.3/23/12.9 | 12.1/1.9/0 |
| Miembros inf. Distal Leve/mod/sev(%) | 14.2/18.3/30.3 | 16.8/6.5/0 |
| Reflejos: Hiperreflexia/Arreflexia(%) | 1.1/89.4 | 10.2/38.9 |
| Alergia inespecifica(%) | 9.4 | 11.1 |
| Depresión (%) | 13.9 | 15.8 |
| Ansiedad(%) | 3.3 | 1.9 |
| Disfunción urinaria(%) | 43.7 | 38.9 |
Traducido y adaptado de Reetz, Dogan y Col. (2018) (66)
DIAGNÓSTICO
Todo paciente con sospecha clínica de AF debería completar una evaluación clínica y neurológica integral. Los exámenes complementarios para AF (neurofisiológicos, imagen y laboratoriales) están orientados a estimar la extensión del compromiso neurológico y sistémico. El diagnóstico definitivo se confirma con un estudio genético.
Los estudios de conducción nerviosa evidencian un compromiso neurofisiológico temprano con reducción o ausencia de potenciales de acción sensitiva; sin embargo, esto podría variar según el fenotipo. Los fenotipos de inicio tardío suelen tener una conducción nerviosa normal 88.
La Imagen por Resonancia Magnética (IRM) cerebral habitualmente no muestra cambios significativos en el cerebelo; sin embargo, se pueden apreciar diversos grados de atrofia del tronco encefálico afectando principalmente la región central del mesencéfalo, los pedúnculos cerebelosos, el vermis rostral, del núcleo dentado, los núcleos de la base, el cuerpo calloso y el quiasma óptico, que se observan sobretodo en estadios avanzados de la enfermedad 18. Por el contrario, la IRM de columna muestra el adelgazamiento de la médula espinal cervical y torácica con disminución de su diámetro anteroposterior esto debido al adelgazamiento de las columnas dorsales y los tractos espinocerebelosos 89.
En la evaluación cardiaca, se recomienda realizar un electrocardiograma y un ecocardiograma en el momento del diagnóstico, y posteriormente con una frecuencia anual para descartar alteraciones estructurales. En pacientes con palpitaciones se sugiere realizar una evaluación con un monitoreo Holter para descartar alteraciones de la conducción cardíaca 86,90.
Respecto de los exámenes séricos, se sugiere realizar el dosaje de glucosa y pruebas de tolerancia oral a la glucosa por lo menos una vez al año para detectar cambios tempranos que permitan un diagnóstico oportuno de diabetes. Otros estudios serológicos como el hemograma, perfil tiroideo, dosaje de vitamina E y B, resultan útiles en el diagnóstico diferencial de la AF.86
Diagnóstico genético
La técnica molecular “gold standard” para la determinación cuantitativa del número de repeticiones del de la expansión GAA en el gen FXN es el Southern Blot; sin embargo, su uso está restringido para la investigación debido a su alto costo y dificultad. Por ello, habitualmente el diagnóstico molecular para AF se realiza con las técnicas PCR y TP-PCR y electroforesis capilar13,91 . En pacientes con sospecha clínica de AF y con un solo alelo expandido, se recomienda el uso de la amplificación de sondas dependiente de ligandos múltiples (MLPA) y/o la secuenciación de los exones del gen FXN, con el fin de determinar deleciones o variantes puntuales91.
A pesar de los avances en el diagnóstico y la introducción de pruebas genéticas persiste un retraso de 4-5 años en el diagnóstico de AF en pacientes con síntomas neurológicos y hasta de 6 años en pacientes sin síntomas neurológicos 62.
Diagnóstico diferencial
El diagnóstico diferencial de la AF es amplio y el interrogatorio, los antecedentes patológicos y exámenes serológicos juegan un rol importante en el diagnóstico. En un paciente con ataxia progresiva esporádica se deben descartar causas secundarias: toxicidad por fármacos incluyendo alcohol, litio, antiepilépticos y quimioterápicos; causas inmunomediadas por formación de anticuerpos (anticuerpos antitiroideos, anti-GAD, ataxia relacionada a gluten), ataxia por degeneración cerebelosa paraneoplásica, ataxias por deficiencia de vitaminas B1 y B12, causas infecciosas y otras enfermedades degenerativas como la atrofia multisistémica 92.
En pacientes con antecedentes familiares, se deben considerar las ataxias dominantes de penetrancia incompleta como algunas ataxias espinocerebelosas. Las ataxias recesivas que se deben considerar en el diferencial incluyen la ataxia por deficiencia de vitamina E, con una presentación muy parecida a la AF, pero suelen tener más compromiso en la agudeza visual y concentraciones bajas de vitamina E en sangre. La Ataxia Telangiectasia que cursa con niveles de alfafetoproteína elevados. Otras ataxias hereditarias para considerar son las ataxias con apraxia oculomotora, ataxia espástica de Charlevoix-Saguenay, xantomatosis cerebrotendinosa, síndrome de Marinesco-Sjögren; estas a diferencia de la AF cursan con atrofia cerebelar temprana. En pacientes con predominio de neuropatías periféricas y pie cavo se plantea como diagnóstico diferencial la enfermedad de Charcot Marie Tooth. Dentro de los desórdenes metabólicos hereditarios que cursan con ataxia podemos encontrar la enfermedad de Wilson por acúmulo de cobre en los ganglios basales; la deficiencia de coenzima Q10 y otros desórdenes mitocondriales 93.
Aproximación terapéutica en AF
El paciente con sospecha de AF debe ser evaluado por un equipo multidisciplinario conformado por especialistas en neurología, cardiología, endocrinología, medicina física, psiquiatría, oftalmología, otorrinolaringología, traumatología y genética médica 86,93.
Recientemente (febrero, 2023), la FDA aprobó el uso de omaveloxolona 150mg/día, un activador del factor transcripcional NFR2- proteína que regula la respuesta ante el estrés oxidativo- como tratamiento farmacológico en AF en mayores de 15 años. Los ensayos clínicos con este fármaco han mostrado una mejoría de la función bulbar, coordinación de las extremidades y estabilidad en sus participantes. El aumento de enzimas hepáticas fue el evento adverso más significativo por lo que se sugiere controles periódicos con dosaje aminotransferasas bilirrubinas séricas. Otros efectos adversos reportados están el aumento de los niveles de colesterol y aumento del Péptido natriurético B, sugiriéndose una evaluación cardíaca para evaluar continuidad del fármaco en estos casos 94-96. Este fármaco aún no se encuentra disponible a nivel regional.
Otros fármacos para AF aún en estudio incluyen idebenona y la Coenzima Q10 que son fármacos con efecto antioxidante. La epicatequina y los agonistas de PPARγ, como la leriglitazona, actuarían mejorando la función mitocondrial. Los inhibidores de la histona deacetilasa, el interferón-gamma, la eritropoyetina (EPO) y el resveratrol actuarían aumentando la actividad de la frataxina 97.
Terapia génica
En los últimos años han incrementado los estudios de terapia génica en AF. Estas terapias consisten en la administración de terapias a través de vectores virales y no virales que ulteriormente codifiquen FXN. Los vectores virales utilizados incluyen lentivirus, el virus del herpes simple tipo 1, adenovirus retrovirus y poxvirus 98. Si bien varios ensayos han mostrado resultados alentadores por su eficiencia de transducción, hay dificultades en la especificidad de los métodos de administración. La sobreexpresión de FXN no está exenta de riesgos y complicaciones como toxicidad cardiaca y hepática 99. Al momento no existe terapia génica alguna aprobada para AF.
Pronóstico
La expectativa de vida de los pacientes con AF promedio es de 40 a 50 años, y los que tienen un mayor compromiso pueden fallecer incluso antes de los 40 años. La principal causa de muerte en AF incluye la cardiomiopatía y síndromes relacionados 74,100).
Perspectivas sobre el diagnóstico y cuidado de AF en Perú
La aplicación universal de las recomendaciones planteadas para el diagnóstico oportuno y el manejo integral de AF tiene grandes retos en el Perú y varias regiones de Latinoamérica. El entrenamiento en genética y enfermedades raras en pre y posgrado es aún insuficiente, y en varias regiones casi inexistente. Esta debilidad en la formación médica asocia una baja sospecha diagnóstica, orientación inadecuada de los casos con exceso de exámenes auxiliares con poco valor predictivo al diagnóstico definitivo. El acceso limitado a tecnologías de genotipificación y secuenciación para enfermedades por expansión de repeticiones como el triplete GAA de Friedreich y más aún secuenciación para identificación de variantes puntuales, limita que casos de AF tengan diagnóstico definitivo. Las limitaciones de acceso directo o referencia efectiva a evaluaciones multidisciplinarias incluyendo medicina física y rehabilitación, cardiología, endocrinología entre otras que usualmente se concentran en las grandes urbes, afectan directamente la calidad de atención y manejo de los casos de AF. La limitada cantidad de especialistas con formación en genética y asesoramiento genético dificulta que los pacientes con AF y familiares en riesgo reciban orientación e información adecuada para la toma de decisiones respecto a planificación familiar, diagnóstico presintomático y otras decisiones vitales. La omaveloxolona recientemente aprobada por FDA para AF, cuenta con autorización para su comercialización, sin embargo aún no está disponible en el mercado y posiblemente sea una alternativa inalcanzable en varias regiones de Latinoamérica por su costo elevado, burocratización de los trámites de autorización de uso y presupuestos gubernamentales que difícilmente pueden priorizar la atención de enfermedades raras o huérfanas.

