











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La enfermedad de Huntington (EH) es un trastorno neurodegenerativo hereditario autosómico dominante, determinada por una tríada de características motoras, cognitivas y psiquiátricas 1. La EH es causada por una expansión anormal del trinucleótido citosina-adenina-guanina (CAG) en una región del exón 1 en el gen HTT o gen de la huntingtina 2. Las personas portadoras de 40 a más repeticiones de CAG presentarán síntomas de la enfermedad en algún momento de su vida, en tanto que aquellas con 36 a 39 repeticiones expresarán la enfermedad con una penetrancia reducida 3. La EH tiene distribución mundial con variaciones geográficas, con una prevalencia mundial aproximada de 5,5 por cada 100 000 personas 4, por ello es considerada como una enfermedad rara o huérfana. En el Perú, la población con un gran porcentaje de EH es el valle de Cañete, que podría ser la segunda población con EH más grande de América Latina y una de las más conocidas a nivel mundial 5.
La mayoría de personas con EH desarrollan síntomas durante la edad adulta, con una edad media de 40 años, en un rango de 2 a 79 años 6. Sin embargo, existen formas de inicio juvenil de la Enfermedad de Huntington (EHJ) antes de los 20 años 7. En 1883, Carl Friedrich Otto Westphal describió el caso de una persona de 18 años con EH que presentaba una rigidez prominente, denominada en adelante como variante de Westphal 8. Los casos de EHJ representan alrededor del 6% de todos los casos de EH a nivel mundial, generalmente se asocia con una expansión de CAG mayor a 60 repeticiones 9. En el Perú, los casos descritos de EHJ cursan con formas parkinsonianas, coreicas, asi como formas infantiles con ataxia, epilepsia y regresión del desarrollo 10.
Los casos con sospecha clínica de EH con estudio genético negativo para la enfermedad se catalogan como fenocopias de EH, y representan alrededor del 1 % de los casos con sospecha clínica de EH 11. Múltiples enfermedades genéticas pueden expresarse clínicamente como fenocopias de la EH, entre las que destacan las enfermedades de Huntington Like (HDL) tipo 1, 2 y 4. La HDL2 es la fenocopia de EH más frecuente en América del Sur 12, y cursa con síntomas cognitivos, psiquiátricos y motores muy similares a la EH. Otras enfermedades hereditarias que cursan con síndrome coreico incluyen la atrofia dentorubropalidoluisiana (DRPLA), la neuroferritinopatía y la corea hereditaria benigna 13. Otras causas secundarias pueden expresarse como fenocopias de EH, como las discinesias asociadas al uso crónico de antipsicóticos.
A continuación presentamos dos hermanos con movimientos involuntarios y problemas del comportamiento, uno con diagnóstico definitivo de EHJ y el otro con fenocopia intrafamiliar. Se destaca la importancia de la evaluación clínica detallada y del estudio genético en todo individuo con sospecha de EH.
CASO 1
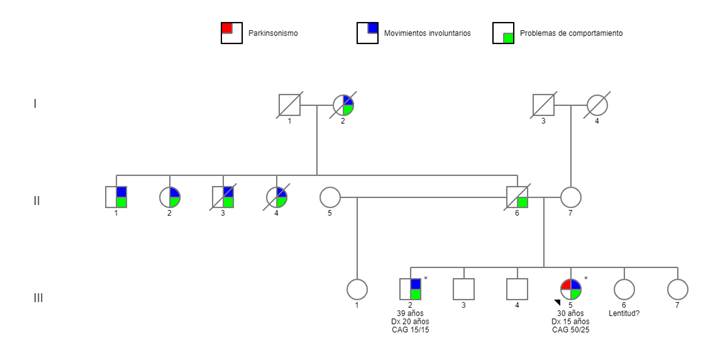
Se trata de una mujer de 30 años (individuo III-5, figura 1), soltera, secundaria incompleta, natural de la sierra central del Perú. El padre, ya fallecido, tuvo antecedente de hábito aparentemente alcohólico, con múltiples parejas, conducta violenta hacia su familia y crianza muy estricta. La paciente inició la enfermedad a los 15 años con lentitud progresiva en los movimientos voluntarios, inquietud psicomotriz nocturna, aislamiento, labilidad emocional y obstinación. Asimismo, tenía dificultad para articular palabras, episodios de alucinaciones y lenguaje incoherente, que incluso ameritaron evaluación psiquiátrica en un hospital de su localidad y otro especializado en salud mental en Lima; para lo cual recibió un tratamiento farmacológico de risperidona.
En el examen neurológico inicial, realizado a los 21 años de edad, se aprecia bradicinesia global con rigidez bilateral y axial, hipomimia moderada, incoordinación de manos y pies, tics vocales, movimientos de seguimiento ocular lentos, latencia incrementada de sacádicos y alteraciones cognitivas. En la escala unificada de valoración de la Enfermedad de Huntington, segmento motor (UHDRS III): 12 puntos; Mini-mental State Examination (MMSE): 12 puntos; escala de la evaluación cognitiva de Montreal (MoCA): 13 puntos; y estudio genético del gen HTT: 50/25 repeticiones de CAG.
Recibió tratamiento farmacológico con risperidona 2 mg/día y alprazolam 0.5 mg/día con buena adherencia y sin efectos secundarios destacables. Tuvo seguimiento regular por aproximadamente seis años con evolución tórpida con parkinsonismo rígido-acinético moderado. Según evaluación, nueve años después de la primera atención se encuentra parcialmente dependiente, poco comunicativa y con caídas aisladas.
CASO 2
Se trata de un varón de 39 años (individuo III2, figura 1), soltero, grado de instrucción superior incompleto, natural de la sierra central de Perú. Inició la enfermedad a los 20 años, aproximadamente con cambios de comportamiento, ideas delusivas y alucinaciones auditivas. Recibió tratamiento en la especialidad de psiquiatría en el hospital de su localidad y otro especializado en salud mental en Lima. Se le diagnosticó esquizofrenia y le recetaron antipsicóticos y otros fármacos, como benzodiacepinas, estabilizadores del ánimo y anticolinérgicos, con respuesta fluctuante al tratamiento a lo largo de los años. No están claros los primeros síntomas que motivaron que esté en un hospital psiquiátrico. A los 29 años, presentó episodio de parpadeo constante referido como ardor, tics faciales, mordedura, inquietud a predominio nocturno e irritabilidad. Entre sus antecedentes personales de importancia, se tiene: convulsión con fiebre a los 7 años, atención en instituto especializado de neurología para descartar epilepsia a los 15 años, impulso sexual incrementado desde los 12 años, con historia de denuncias por hostigamiento sexual a mujeres, acudía a prostíbulos desde los 18 años y tenía relaciones sexuales con otros hombres. Tuvo un intento suicida a los 26 años. Ha laborado ocasionalmente por períodos cortos en tareas sencillas, como recolección de café en una granja.
A los 30 años, presenta movimientos anormales (tipo «sacudidas») en extremidades y tics en el rostro, incoordinación de manos y pies e inestabilidad de la marcha, conducta desorganizada, como encender y apagar la luz en forma repetida, risas inmotivadas y conducta agresiva hacia familiares. Recibió valproato de sodio 500 mg/día por sospecha de crisis epilépticas vs. pseudocrisis, y biperideno 4mg/día para prevención de síntomas secundarios. Posteriormente, se modificó el tratamiento farmacológico y se le receta risperidona y clonazepam, cuyas dosis llegaron a ser de 5 mg/día y 1,5 mg/ día, respectivamente, en consultas de seguimiento subsecuentes. Recibió ciclos cortos de otros antipsicóticos, como clorpromacina, olanzapina y quetiapina, sin obtener mejoría significativa.
Al examen neurológico inicial, realizado a los 30 años de edad, se aprecia disfagia, disartria, tics vocales, movimientos coreicos en rostro y miembros superiores de tipo constante y de poca amplitud, movimientos distónicos cervicales con desviación del cuello hacia a la izquierda y de tronco, rigidez en miembros superiores con predominio derecho. En UHDRS III: 13 puntos; en MMSE: 25 puntos; y en MoCA: 19 puntos. El estudio genético inicial en el gen HTT resultó no informativo por presencia de alelo único en el gen HTT; un estudio genético posterior, mediante Repeat primed Polymerase Chain Reaction (RP-PCR), confirmó el genotipo de 15/15 repeticiones de CAG en el gen HTT, siendo negativo para EH.
La evaluación psiquiátrica realizada a los 33 años describe al paciente con lentitud motora, movimientos distónicos e involuntarios generalizados, lenguaje con tono adecuado, ánimo aplanado, orientado en tres esferas, delusiones autorreferenciales y de contenido sexual, significados adventicios, alucinaciones auditivas, tendencia a la irritabilidad, desinhibición sexual, comportamiento sexual inadecuado orientado hacia mujeres y heteroagresividad, sin conciencia de la enfermedad.
Tuvo seguimiento regular por aproximadamente seis años con mala respuesta al tratamiento de los movimientos involuntarios y distonía cervical, pero con preservación de las funciones cognitivas. Según la evaluación, nueve años después de la primera atención, se encuentra lúcido, con remembranza del pasado, leve disartria, movimientos involuntarios y distonía severa con predominio cervical y miembros superiores. En opinión del familiar responsable, hay control parcial de los trastornos del comportamiento y muy poco control de los movimientos involuntarios.
DISCUSIÓN
Presentamos dos casos con un fenotipo compatible con EH. El estudio genético confirmó el diagnóstico de EH en la probando (individuo III-5). Sorprendentemente, el hermano (individuo III-2), con un estudio genético inicial incierto, tuvo un resultado genético confirmatorio negativo para EH, definiéndose como una fenocopia intrafamiliar de EH.
La probando (III-5) con EHJ, desde los 15 años cursa con un parkinsonismo bilateral y simétrico de predominio rígido-acinético asociado a cambios progresivos de comportamiento, fenotipo compatible con la variante de Westphal de EH. Esta variante es más frecuente en individuos que inician síntomas antes de los 21 años 14. El genotipo de la probando es 50/25 repeticiones de CAG y se presenta con una forma temprana de la enfermedad, concordante con lo descrito en la literatura, en la que se menciona que a más repeticiones la enfermedad se presenta en edades más tempranas, fenómeno conocido como anticipación génica 15. Se postula que el parkinsonismo en EHJ ocurriría por una pérdida neuronal progresiva del putamen y núcleo caudado en un cerebro aún en maduración 16.
El diagnóstico de fenocopia intrafamiliar del hermano (III-2) fue tardío. Debido a que la técnica que se utilizó inicialmente no diferenciaba el homoalelismo de la presencia de un genotipo con alelo de expansión grande no visible (más de 40 repeticiones de CAG), se asumió que era un caso probable de expansión grande, dado que era hermano de un caso ya diagnosticado con EH. El diagnóstico genético negativo definitivo se produjo luego de un segundo análisis con RP-PCR, que confirmó homoalelismo en el gen HTT con 15 repeticiones de CAG. Los movimientos involuntarios de predominio cervicofacial, de aparición posterior al trastorno de comportamiento, estarían relacionados con una discinesia tardía secundaria al uso prolongado de neurolépticos 17. Considerando la sospecha de crisis epilépticas, los síntomas psiquiátricos y motores, no podemos descartar la coexistencia de alguna enfermedad neurodegenerativa que se exprese como fenocopia de la EH, incluyendo el HDL2 18, entre otras que no fueron sistemáticamente descartadas en este caso.
Las conductas de desinhibición sexual a edad temprana, la conducta hipersexual e incluso los intentos de agresión sexual pueden indicarnos la presencia de un trastorno psiquiátrico a edad temprana, que puede diagnosticarse como un trastorno parafílico e hipersexualidad. Estos diagnósticos son más frecuentes en varones en el inicio de la adolescencia. Usualmente, se da el diagnóstico de uno o más trastornos parafílicos, además de comorbilidades psiquiátricas como psicosis 19. Los trastornos parafílicos y la hipersexualidad pueden ser parte de una enfermedad neurológica, sea dada por una lesión cortical o subcortical del cerebro. Sin embargo, en este caso, los síntomas de la conducta sexual anteceden al desarrollo de los primeros síntomas psicóticos (20, 21). En el caso de las psicosis, es posible considerar un diagnóstico de esquizofrenia. De acuerdo con el Manual de diagnóstico y estadístico de los trastornos mentales (DSM-5) 22, el individuo (III-2) cumple con la presencia de síntomas compatibles, como delusiones, alucinaciones, conducta desorganizada, disminución de la motivación (apatía), así como la alteración significativa de su funcionamiento en sus relaciones interpersonales, laborales o de autocuidado.
En conclusión, la EH puede expresarse con fenotipos distintos dependiendo de la edad de inicio, y ello incluye formas atípicas de parkinsonismo en menores de 20 años. Las fenocopias de EH pueden presentarse incluso dentro de una familia con Huntington confirmado, como ocurrió en este caso. Se recomienda que cada caso dentro de una familia tenga una evaluación completa con estudio genético independiente, además de un diagnóstico definitivo que permita un asesoramiento y seguimiento adecuado para cada miembro afectado.

