










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Medica Herediana
versión impresa ISSN 1018-130X
Rev Med Hered v.6 n.3 Lima abr. 1995
Incontinentia pigmenti: A propósito de un caso.
Incontinentia pigmenti.
Tori Tori Carlos*. Villar De Cipriani Enriqueta**, Arias Stella, Javier***, Avalos Carmen****.
*Pediatra, Clínica Ambulatoria, Universidad Peruana Cayetano Heredia y Clínica San Felipe. Lima, Perú.
**Médico Dermatólogo, Clínica Médica Ambulatoria, Universidad Peruana Cayetano Heredia. Lima, Perú.
***Profesor Principal, Departamento de Patología, Universidad Cayetano Heredia y Clínica San Felipe. Lima Perú.
****Médico Oftalmólogo, Centro Oftalmológico, Miraflores. Lima, Perú.
SUMMARY
Incontinentia Pigmenti is a rare X-linked multisystem neuroectodermic disorder with signs and symptoms related mainly to the dermatologic, dental, ocular and central nervous systems, and characterized by death in the majority of male embryos. Affected children do not appear sick, in spite of the skin eruption, the peripheral leucocytosis and marked eosinophilia. Most of the cases are reported in caucasians, although there are description in black children, orientals, north central and south american Indians, and in our mestizos. The name of Incontinentia Pigmenti describes the incontinence of the melanin pigment from the basal layer of the epidemermis into the superficial dermis.
KEY WORDS: Incontinentia Pigmenti, children, melanin, Bloch Sulzberger, neuroectodermic.
INTRODUCCIÓN
La Incontinentia Pigmenti (IP), es un sindrome neuroectodérmico mulsistémico, de rara ocurrencia, con herencia dominante ligada al cromosoma X, que se acompaña de signos dermatológicos, oculares, dentales, del sistema nervioso central y caracterizado por la muerte de la mayoría de los embriones masculinos (1). Los niños generalmente no lucen enfermos, a pesar de la erupción, la leucocitosis y la marcada eosinofilia (2). La mayoría de casos son de raza blanca, pero ocurren en niños de raza negra, oriental, indios norteamericanos, centro y sudamericanos, y en nuestra raza mestiza.
Caso clínico
S.A.M. mujer, de cinco semanas de edad, nació con 5 "ampollitas" en el antebrazo izquierdo, y otra en la cara dorsal de la tercera falange del dedo medio de la mano derecha. Tres días después aparecieron mácula eritematosas en las extremidades y vesículas pequeñas en los flancos del abdomen, que se pigmentaron diez días después( Figura Nº1 y figura Nº2). Algunas era lineales, la mayoría tortuosas, serpiginosas, otras en mancha. Habían pápulas liquenoides en el antebrazo derecho. Todas ellas respetaban la línea media, salvo en la región lumbosacra. Tenía una lesión verrucosa, de 3 mm de diámetro en el pliegue proximal de la región dorsal de la tercera falange del dedo medio derecho.
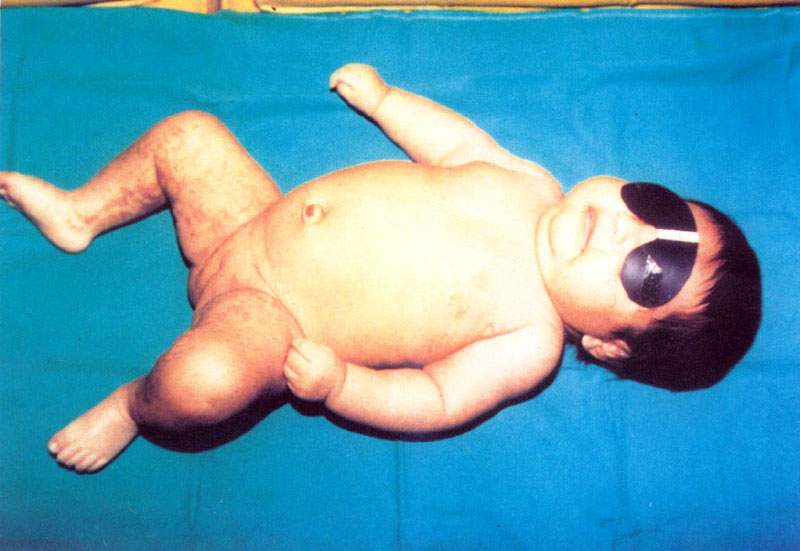
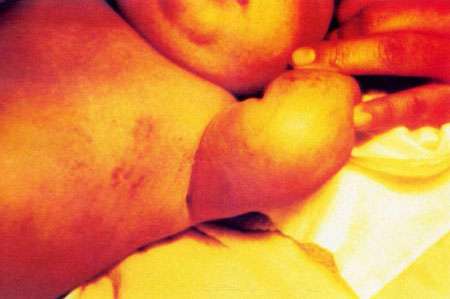
Antecedentes Familiares
Tía paterna de 12 años de edad, con convulsiones. Nadie en la familia presenta o ha presentado las lesiones características de esta entidad.
Anatomía Patológica
En las zonas pigmentadas se aprecian las capas queratínicas y granulosas. Ocasionales células disqueratónicas en la capa espinosa. Algunas células con pigmento melánico en la capa basal ( Figura Nº3). Presencia de melanóforos perivasculares en la dermis superficial e infiltración leucocitaria en algunos folículos ( Figura Nº4).
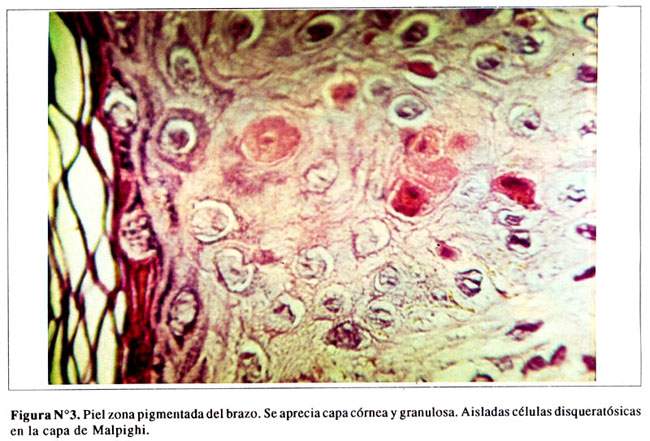
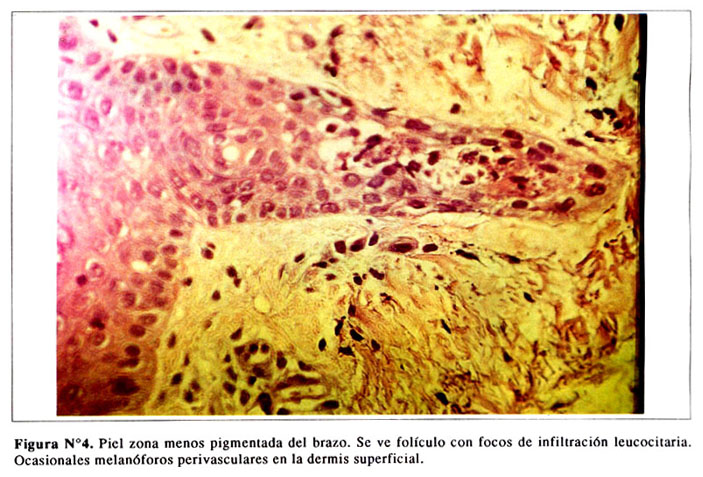
DISCUSION
La IP es una enfermedad hereditaria, ligada a X, dominante, con letalidad en los varones. Las mujeres afectadas tienen un alto índice de abortos espontáneo de fetos varones (3).
Se han identificado hasta el momento dos genes en el cromosoma X relacionados con esta enfermedad. La región involucrada ha sido siempre la misma: Xp 11; ese gen se ha llamado IP 1. Todos los casos en los que el gen IP 1 ha estado involucrado han sido esporádicos. En los casos familiares se ha encontrado una asociación con un gen situado en el brazo largo de X, en la región Xq28 y se denomina IP 2.
En el Perú hay dos casos publicados de Incontinencia Pigmento (4,5).
Estas enfermedad comprende la piel en un 98%, las uñas en un 40%, el pelo en un 50%, los dientes en el 65 a 80%, los ojos en el 35 a 40%, el sistema nervioso central en 30% y la glándula mamaria (2,3,6,7).
En el 90% de casos, la erupción se inicia en la primeras dos semanas de vida, y ocurren cuatro estadíos bien definidos.
El estadío 1 a menudo se confunde con exantemas virales o estafilococias de piel y son máculas eritematosas seguidas de vesículas, siguiendo un patrón lineal al predominio de la cara flexora de las extremidades, y caras laterales del tronco, respetando la cara y la línea media.
El estadío 2 se presenta en un 80% de casos (3), aparece entre la segunda y sexta semana de edad (8). Son lesiones típicamente hiperqueratósicas y verrucosas localizadas sobre la superficie dorsal de los dedos de las manos y pies, y en las regiones axilares e inguinales (2,5,8).
El estadío 3 es característico, y ocurre en el 98% de casos (2,6). Se inicia entre los 3 y 6 meses de edad; la pigmentación es gradual y ocurre después que las vesículas han desaparecido. Su apariencia es estriada o en torbellino con respecto a las líneas de Blaschko y son más frecuentes en el tronco que en las extremidades. Entre el 14% y el 40% de pacientes con IP se presentan en esta fase, sin evidencia de los estadíos anteriores (2,9).
En el estadío 4 hay áreas circulares o lineales sin pelo, pálidas, y a predominio de los miembros inferiores (10). Puede haber distrofia ungueal y tumores subungueales keratóticos (11), asociado a deformidad óseas de las falanges subyacentes. El pelo es grueso, sin brillo, y erizado (12), con alopecia en el vertex (2). Se describen pezones supernumerarios, hipoplásicos y con pigmentación anormal, e hipoplasia o aplasia de la glándula mamaria (10). Puede haber aplasia parcial de las glándulas sudoríparas (13). La dentición decidua y permanente puede estar afectada en un 80% presentando anodoncia, hipodoncia, erupciones tardías impactación y malformación de las coronas (11,14). El sistema ocular puede estar comprometido y es uno de los que incide en el pronóstico de esta enfermedad. Las lesiones oftalmológicas pueden afectar la reina y áreas no retinales (6,15). En la retina pueden ocurrir zonas de isquemias que promueven la proliferación de neovasos sanguíneos, con sangrado y fibrosis subsecuentes, algo similar a lo que se describe en la retinopatía del prematuro (6,16). Las no retinales incluyen el estrabismo, el microftalmus, la pigmentación conjuntival (17), las cataratas congénitas (2), la uveitis, la atrofia del cuerpo ciliar, la ausencia de cámara anterior, la atrofia del nervio óptico y las escaleras azules (2,8,18). En nuestro paciente no se observaron lesiones oculares.
El compromiso del Sistemas Nervioso Central afecta también seriamente el pronóstico de la incontinencia pigmento, sobretodo si ocurre en el período neonatal. Lo más frecuente son las convulsiones, la parálisis espástica, el retardo psicomotor, la microcefalia, la necrosis hemorrágica y la hidrocefalia (2).
La incidencia de retardo mental severo en casos familiares es del 3%, comparado con un 15 % en casos esporádicos de IP (3).
El 13.7% de los pacientes con IP presentan deformidades del tronco, enanismo, espina bífida, paladar hendido, hemiatrofia, luxación congénita de cadera y condrodistrofia (17,18).
REFERENCIAS BIBLIOGRAFICAS
1. Lenz W. Zur Genetic der Incontinentia pigmenti. Ann Paediatr 1961; 196: 149-65. [ Links ]
2. Carnev RG. Incontinentia pigmenti: A world statistics análisis. Arch Dermatol 1976; 112: 535-42. [ Links ]
3. Landy SJ, Donnai D. Incontinentia Pigmenti. J Med Genet 1993; 30: 53-59. [ Links ]
4. Luy M, Noriega E, Llosa G. Incontinentia Pigmenti. Sindrome de Bloch-Sulzberger. Comunicación de un caso. Revista Peruana de Pediatría 1963; 21: 144-49. [ Links ]
5. Arana G, Ricse H, Small O. Incontinentia pigmenti clásica. Tribuna Médica 1976; 40(473): 30-2. [ Links ]
6. Goldberg MF, Curtis PH. Retinal and other manifestations of incontinentia pigmento. Ophthalmology 1993; 100: 1645-54. [ Links ]
7. Catalano RA. Incontinentia Pigementi. American Journal of Ophthalmology 1990; 110: 696-700. [ Links ]
8. Cohen B. Incontinentia pigmenti. Neurologic Clinics 1987; 5: 361-77. [ Links ]
9. Felt SE, Jacobs DE. Incontinentia pigmenti J Kan Med Soc 1973; 74: 43-5. [ Links ]
10. Moss C, Ince P. Anhydrotic and achromians lesions in incontinentia pigmenti. Br J Dermatol 1987; 116: 839-50. [ Links ]
11. Simmons DA, Kegel MF, Scher RK, Hines YC. Subungual turnous in incontinentia pigmenti. Arch Dermatol 1986; 122: 1431-4. [ Links ]
12. Wiklund DA. Incontinentia pigmenti. A four generation study. Arch Dermatol 1980; 116: 701-3. [ Links ]
13. Ramos N. Incontinentia pigmentaria. Tesis de Bachiller. Lima, Perú. Universidad Peruana Cayetano Heredia, 1988. 107 pp . [ Links ]
14. Niccoli-Filho WD, Da Rocha JC, Di Nicolo R, Sraindarian PI. Incontinentia pigmenti. a case report. J Clin Pediatr Dent 1993; 17: 251-3. [ Links ]
15. Follew SM, Greenwald MJ, Prendiville JS. Jampol LM. Ocular findings of incontinentia pigmenti in a male infant with Klinefelter syndrome. J Pediatr Ophthalmol Strabismus 1992; 29: 180-4. [ Links ]
16. Fracois J. Incontinentia pigmenti (Bloch-Sulzberger syndrome) and Retinal changes. Brit Journal of Ophthalmology 1984; 68: 19-25. [ Links ]
17. Mc Crary JA III. Smith JL. Conjuntival and retinal incontinentia pigmenti. Arch Ophathalmol 1968; 79: 417-22. [ Links ]
18. Watzke RC, Stevens TS, Carney RG Jr. retinal vascular changes of incontinentia pigmenti. Arch Ophtalmol 1976; 94: 743-6. [ Links ]
Correspondencia:
Carlos Tori Tori
Centro Médico San Felipe.
Av. Gregorio Escobedo 660, Jesús María
Lima, Perú.
