










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Medica Herediana
versão impressa ISSN 1018-130Xversão On-line ISSN 1729-214X
Rev Med Hered v.15 n.3 Lima jul./set. 2004
CFTR: 15 años después del descubrimiento de un gen
CFTR: 15 years after a gene discovery
Vega-Briceño, Luis Enrique *
*Hospital Clínico Universidad Católica de Chile
INTRODUCCIÓN
La fibrosis quística (FQ) es la enfermedad autosómica recesiva letal más común en la población blanca del Reino Unido, con una incidencia estimada en 1/2000 recién nacidos vivos (1). En los Estados Unidos, el 50% de los pacientes con FQ son diagnosticados a la edad de 6 meses y el 90% a la edad de 8 años (2). La edad promedio de sobrevida estimada en niños Europeos nacidos en la década del 90 se calcula en 40 años, lo cual representa el doble de lo que fué en los últimos 20 años; en donde sólo la mitad de ellos son ahora adultos (3). Definitivamente, este incremento en la sobrevida es el resultado de un buen trabajo en el área de la fisioterapia, nutrición y un tratamiento antibiótico agresivo; y por supuesto un incremento considerable del entendimiento de ésta enfermedad. El presente artículo es una revisión de algunos aspectos de la fisiopatología, microbiología y terapéuticos de la FQ, al celebrarse el 15 aniversario de la clonación del gen y su vez es una actualización del último artículo publicado en esta misma revista (4).
Fisiopatología
La clonación de un gen
El más importante paso en el proceso de entendimiento de la FQ fue sin lugar a dudas la clonación del gen en el año 1989 (5, 6). Localizado en el brazo largo del cromosoma 7 y codificado como CFTR, por sus siglas en inglés; es una proteína con 1480 amino ácidos situada en la porción apical de la membrana de las células epiteliales, que se expresa en las células secretorias, senos paranasales, pulmones, páncreas, hígado y tracto reproductivo (7). Consiste en doce regiones de membranas hidrofóbicas ligadas a dos puentes nucleótidos con su respectivo dominio conteniendo múltiples sitios para la fosforilación. Desde que el gen fue clonado, 1291 mutaciones han sido ya descritas e identificadas en el mundo (8).
La forma más frecuente de mutación es la delección del codón que produce la pérdida de un residuo de fenilalanina en la posición 508, denominada mutación DeltaF508. Está presente en cerca del 70% de todos pacientes con FQ, aunque existen grandes variaciones geográficas descritas con rangos desde 32-82% (5, 9). En Chile por ejemplo, Navarro y colaboradores reportaron que la prevalencia de ésta mutación representaba el 50% de las mutaciones en FQ (10) siendo la segunda mutación más frecuente la G542X (11).
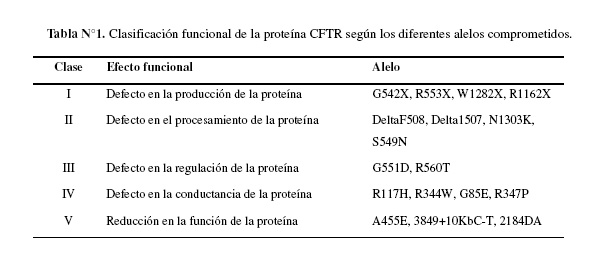
Existen cinco clases de mutaciones descritas (Tabla N°1). La mutación Clase I, resultante de un defecto de inestabilidad del ácido ribonucleico mensajero (mRNA) o de una proteína anormal, la cual es rápidamente degradada. Las mutaciones Clase II resultan de una falla en el proceso de síntesis de la proteína o del transporte de la misma a través de la membrana apical de la célula. Esta clase incluye a la mutación DeltaF508. La mutación Clase III, resulta de una proteína correctamente localizada pero defectuosa en la regulación de la actividad del canal de cloro. La mutación Clase IV, está correctamente localizada y regulada pero tiene un defecto en la conductancia del cloro y finalmente la mutación Clase V, resulta en una reducida síntesis de CFTR, con una clara tendencia a presentar una enfermedad leve, con test de sudor en valores límites y con una secresión intestinal residual de cloro (12).
Las manifestaciones clínicas de la enfermedad varían mucho entre individuos afectados pero se reconoce que las mutaciones clases 1-3 son las más comunes, estando asociadas usualmente con insuficiencia pancreática (13). Las diferencias en los fenotipos están relacionadas con el efecto de la mutación en la producción de la proteína CFTR; así por ejemplo, valores <3% de producción se asocian a fenotipos más graves (14). Existen factores ambientales o quizás otros genes diferentes a CFTR que también modifican la progresión y la severidad de ésta enfermedad (15).
El rol de CFTR en la patología pulmonar
Varios mecanismos han sido propuestos para explicar como las anormalidades de CFTR pueden predisponer al daño de la vía aérea. La estructura de CFTR muestra homología con el transportador ABC, un exportador dependiente de ATP de macromoléculas desde el interior hacia el exterior de la célula. Hoy en día se acepta que CFTR es un canal de cloro, que ha mostrado ser un inhibidor o autoregulador de los canales de sodio sensibles (7) además de regular la rectificación de la molécula de él mismo (16). El transporte de iones a través de la porción apical de la membrana genera diferenciales de potenciales (DP) que puede ser medido in vivo en la nariz y en la vía aérea baja.
El transporte de sodio predomina en la vía aérea del ser humano y es finalmente el responsable de la negatividad de DP a través del epitelio respiratorio. Este transporte puede ser reducido a niveles normales con el empleo de bloqueadores de canales de sodio, como el amiloride, sugiriendo que la "hiperabsorción" de sodio está incrementada dos o tres veces más en los pacientes con FQ (17), lo cual sugiere que el defecto en el sodio es secundario al defecto del cloro. Se ha confirmado que existe una secresión anormal de cloro en la vía aérea baja de niños con FQ (18) y además se cree que CFTR también regula el transporte de bicarbonato a través de la membrana celular (19).
El transporte de electrolitos es parcialmente responsable de la cantidad y de la composición del fluido en la superficie de la vía aérea. El esputo espeso que se ve típicamente en los pacientes FQ puede ser el resultado de una pobre hidratación del fluido que recubre la superficie de la vía aérea secundario a una secresión anormal de cloro y a una hiperabsorción del sodio. Los tapones mucosos resultantes, impiden la limpieza ciliar normal y por lo tanto la sobrecolonización con bacterias, las cuales inducen una respuesta inflamatoria responsable de la destrucción final del tejido pulmonar. Se ha visto en modelos in vitro, que las altas concentraciones de cloro y sodio inactivan la beta-defensina-1 humana; un péptido natural presente en la superficie de la vía aérea (20).
La composición del fluido de la vía aérea es aún materia de controversia. Algunos investigadores han reportado que no existe diferencia en la composición de la sal entre los sujetos con FQ y los normales, aunque estos experimentos exhiben un sesgo por la forma de recolección de las muestras (21). Se ha sugerido que CFTR puede actuar por si mismo como un receptor de ligason y endocitosis contra P. aeruginosa (22) función que estaría perdida en los pacientes con FQ; por otro lado, existe una falta de regulación en la ligazón de bacterias comúnmente aisladas en sujetos con FQ incrementándose el número de bacterias adherida al epitelio (23).
Tanto P. aeruginosa y S. aureus exhiben un fenotipo mucoide en los pacientes con FQ. La pérdida de agua incrementa la viscosidad del moco y dificulta el clearence mucociliar y el reflejo de la tos. Las bacterias que invaden el pulmón son atrapadas en este moco espeso en el borde apical de la célula en donde encuentran un medio microaerófilo con condiciones favorables para crecer. Esto gatilla un cambio en P. aeruginosa y S. aureus de la forma no mucoide a la forma mucoide, aunque otros estímulos como el peróxido de hidrógeno, también pueden activar este cambio (24,25).
Infección versus inflamación: el debate
Aunque existen estudios que sugieren que un niño con FQ nace con los pulmones normales y sanos (23), Khan y colaboradores demostraron un incremento en el número de neutrófilos y de interleukina 8 (IL-8) en bebes tan jóvenes como 4 semanas sin ninguna evidencia de infección previa (24). Otros estudios por su parte, sugieren que es la infección quien precede a la inflamación (26). La asociación entre CFTR anormal e inflamación se origina con los hallazgos de una IL-10 inhibida aún después de erradicada la infección (27), lo cual a su vez inhibe la producción de IL-1, IL-8 y el factor de necrosis tumoral por las células inflamatorias. Se acepta que una disminución de los niveles de producción de IL-10 por las células epiteliales, puede hacer más susceptible a la célula a una excesiva respuesta inflamatoria ante la infección (28).
Microbiología y uso de antibióticos
En los últimos 10 años ha existido un mejor entendimiento de los patógenos que frecuentemente habitan la vía aérea de los niños con FQ. Staphylococcus aureus y Haemophilus influenza son las bacterias más frecuentemente aisladas en estos niños (7). Resulta interesante comentar que son pocas las enfermedades que muestren una clara distribución de gérmenes adquiridos en función de la edad. La mayoría de estos gérmenes son oportunistas, siendo la excepción S. aureus quien es el único patogénico aún en niños inmunocompetentes.
S. aureus es usualmente es el primer patógeno aislado de la vía aérea de los niños con FQ, por lo que algunos centros Europeos recomiendan una terapia profiláctica antiestafilocócica una vez hecho el diagnóstico de FQ, bajo la premisa que esto mejora la progresión clínica en los primeros dos años de vida (29,30). Contradictoriamente, dos recientes metanálisis no demostraron un claro beneficio al avaluar la profilaxis versus el tratamiento intermitente anti estafilocócico (31,32). Incluso hay estudios que demuestran que la adquisición de Pseudomona se incrementa con el empleo de terapias antiestafilocócicas (33, 34). La revisión sistemática del grupo Cochrane publicada recientemente concluyó que no existe evidencia que soporte esta conducta (32).
Se reconoce que la tasa de infección por S. aureus en FQ disminuye con la edad; sin embargo la tasa de infección por Pseudomona aeruginosa se incrementa (3) Aún con el empleo de nuevas terapias antibióticas agresivas, la infección por la forma mucoide de P. aeruginosa no puede ser erradicada, probablemente por la pobre penetración del antibiótico dentro de los tapones de moco. La infección crónica por P. aeruginosa es reducida en aquellos niños que reciben un tratamiento agresivo con ciprofloxacino oral y nebulización con aminoglicósidos durante las exacerbaciones agudas (35). Hoy en día es frecuente tratar agresivamente el primer aislamiento de Pseudomona con colomicina, tobramicina o gentamicina nebulizada además de algún antibiótico por vía oral o intravenosa. Un panel de expertos de Europa recomienda el uso regular de antibióticos endovenosos cada tres meses independiente del estado respiratorio (36), a raíz de la experiencia danesa quienes reportaron una disminución en la caída de la función pulmonar y mejora en la sobrevida (37); sin embargo ésta no es una estrategia aprobada en otros centros argumentando que no confiere mayor beneficio (38,39).
La Burkholderia cepacia (formalmente Pseudomonas cepacia) emergió en 1970 como un patógeno en FQ (40). Debido a la evidencia de infección cruzada y el terrible impacto en la función pulmonar, todos lo centros tienden a separar a aquellos pacientes colonizados con esta bacteria (41). Otro nuevo organismo es la Stenotrophomonas maltophilia (antes Xanthomonas maltophilia), sin embargo la evidencia actual muestra que la colonización con esta bacteria aparentemente no tiene un impacto en la sobrevida (42). Otras bacterias emergentes como Burkholderia pseudomallei han sido recientemente reportadas en zonas de Tailandia y Australia (43).
A finales de la década pasada, nuevos antibióticos y nuevas rutas de administración han sido estudiados. Ramsey y colaboradores evaluaron 300 mg nebulizados de tobramicina, dos veces al día, mostrando un incremento en el volumen espiratorio forzado en el primer segundo (VEF1) de 10% luego de 5 meses de seguimiento, sin embargo el costo de ésta droga, limitó mucho su empleo (44). El desarrollo de los dispositivos de polvo seco para la entrega de antibióticos aún está en proceso, con la esperanza de brindar un tratamiento de corto tiempo (45). Los nuevos sistemas de nebulización con respiración asistida como el Ventstream y el Pari LC, incrementan la entrega de droga y reducen el tiempo de tratamiento siendo probablemente una promesa.
Inflamación y esteroides
Como se discutió en líneas anteriores, la destrucción pulmonar es el resultado de un círculo vicioso entre infección e inflamación, siendo la gran destrucción tisular producida por la respuesta del huésped frente a los productos generados por P. aeruginosa. Resulta lógico pensar entonces que se podría reducir este daño empleando alguna forma de inmunosupresión. El estudio original empleando prednisona por días alternos fue alentador (46), sin embargo posteriormente se reportó una serie de efectos adversos, como el retraso en la velocidad de crecimiento sin una ganancia significativa en la función pulmonar (47). El uso de esteroides inhalados podría reducir estos efectos indeseables, por lo que son prescritos en más del 40% de los centros de Europa (48); sin embargo aún no existe alguna evidencia sólida que soporte esta conducta. Un estudio denominado CFWISE próximo a ser publicado responderá ésta pregunta; por lo pronto, parece que el uso de esteroides inhalados no incrementa el riesgo de adquirir P. aeruginosa (49).
Ibuprofeno
El Ibuprofeno ha demostrado que inhibe la migración, adherencia y agregación de los neutrófilos. Konstan y colaboradores trataron a pacientes con FQ con altas dosis de ibuprofeno durante 4 años en un estudio bien diseñado, demostrando un menor deterioro de la función pulmonar en el grupo tratado (50). Sin embargo, los niveles séricos de ésta droga necesitan ser controlados estrechamente ya que desde el punto de vista teórico, incluso los niveles incorrectos pueden deteriorar el pulmón (51). Existe una gran variabilidad entre pacientes en las dosis requeridas para alcanzar los niveles adecuados, por lo que la combinación con otras drogas nefrotóxicas, como aminoglicósidos, puede causar insuficiencia renal. Aún se requieren más estudios para determinar su seguridad y eficacia.
Macrólidos: más que un antibiótico.
Existe una fuerte similitud entre FQ y la panbronquiolitis difusa (DPB), una enfermedad encontrada primariamente en orientales (52). Los macrólidos como la eritromicina y azitromicina han sido empleados con gran éxito en la DPB mostrando una reducción significativa en síntomas e incremento en la sobrevida a 10 años en hasta el 90% de los pacientes colonizados con P. aeruginosa (53). Existe evidencia tanto in vitro como in vivo que muestra que éstas drogas exhiben una propiedad antiinflamatoria a dosis mas bajas que su concentración mínima inhibitoria (50). Algunos estudios clínicos soportan la evidencia en FQ, observando un incremento del 5.4 (11%) del VEF1 y la capacidad vital forzada en niños y adultos tratados diariamente con azitromicina por períodos de 4-6 meses (54-56), requiriendo menos cursos antibióticos orales y sin efectos adversos.
Fisioterapia
Una buena fisioterapia es uno de los elementos más importantes para el control de un paciente con FQ. Varios sistemas como los flutter, casacas oscilatorias y ultrasonido han sido desarrollados en los últimos años con la intención de incrementar el clearance de esputo en estos pacientes (57). Un gran avance lo constituye la introducción de las maniobras de espiración forzada como parte de las técnicas de respiraciones cicladas controladas, drenajes autogénicos y el uso de altas presiones al final de la espiración; sin embargo aún no se ha demostrado cual de éstas es superior. Dos completas revisiones en torno al tema han sido publicadas recientemente (58,59).
ALGUNAS NUEVAS TERAPIAS
Mucolíticos
La degradación de los neutrófilos resulta en un incremento en la cantidad de DNA, el cual incrementa la viscosidad del moco. Después de la clonación de la enzima DNAsa en 1990, muchos estudios clínicos evaluaron su seguridad y eficacia reportando una mejoría en la función pulmonar y una reducción en el número de exacerbaciones respiratorias (60, 61). La administración de DNAsa por aerosol, rompe las uniones de DNA en el esputo, disminuyendo la viscosidad del moco y facilitando su eliminación; sin embargo existe gran variabilidad en la respuesta entre diferentes sujetos, con incluso deterioro de la función pulmonar en un tercio de los pacientes (62). El costo de ésta droga en Europa oscila entre 3800 y 7000 Euros/paciente/año para días alternos o continuos respectivamente. La administración de salino hipertónico nebulizado mostró igualmente un incremento de la función pulmonar siendo considerada como una buena alternativa en aquellos que no responden a DNAsa (63).
Terapia genética
Uno de los más grandes avances en la FQ ha sido el desarrollo de la terapia genética en ratones (64). Sólo se requiere de un 10% de expresión de CFTR para generar un transportador normal (65). El principio básico de este tipo de terapia es la inserción de una copia de ADN doble cadena, que codifique un CFTR normal dentro de las células respiratorias en las cuales existe ya un defecto. Para incrementar la tasa de transferencia se emplea la incorporación de un vector como transportador, siendo los adenovirus extremadamente efectivos, sin embargo inducen una respuesta inflamatoria en el huésped que limitan su aplicación clínica (66).
Transplante pulmonar
El primer transplante cardiopulmonar realizado en 1985 (67), abrió una opción terapeútica para aquellos pacientes en estadio terminal. La sobrevida a dos años en aquellos con un VEF1< 30% es mayor del 50% (68), con mejores resultados en adultos que en niños. Lamentablemente, el síndrome de bronquiolitis obliterante continúa siendo la mayor causa de mortalidad, seguido por la presencia de infecciones y el rechazo frente al donante. Hasta comienzos de éste nuevo siglo se habían transplantado más de 1000 pacientes con FQ en el mundo (69); lamentablemente, la demanda de órganos excede la oferta, siendo éste uno de los mayores problemas.
REFERENCIAS BIBLIOGRÁFICAS
1. Dodge JA, Morison S, Lewis PA, Coles EC, Geddes D, Littlewood JM, Scott MT. Incidence, population and survival of cystic fibrosis in the UK, 1968-95. Arch Dis Child 1997;77:493-496. [ Links ]
2. Cystic fibrosis Foundation. Cystic fibrosis Foundation Patient Registry Annual Report 2000. Bethesda: Cystic Fibrosis Foundation, 2001. [ Links ]
3. Elborn JS, Shale DJ, Britton JR. Cystic fibrosis: current survival and population estimates to the year 2000. Thorax 1991;46:881-885. [ Links ]
4. Franchi L. Fibrosis Quística. Rev Med Hered 1994;5:91-96. [ Links ]
5. Rommens JM, Iannuzzi MC, Kerem B-S, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, Zsiga M, Buchwald M, Riordan JR, Tsui L-C, Collins FS. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989;245:1059-1065. [ Links ]
6. Riordan JR, Rommens JM, Kerem B-S, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou J-L, Drumm ML, Iannuzzi MC, Collins FS, Tsui L-C. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989;245:1066-1073. [ Links ]
7. Ratjen F, Doring G. Cystic fibrosis. Lancet. 2003;361:681-9. [ Links ]
8. http://www.genet.sickkids.on.ca/cftr/ ( Visionado el 10 de Enero del 2004).
9. Morral N, Bertranpetit J, Estivill X, et al. The origin of the major cystic fibrosis mutation (delta F508) in European populations. Nat Genet 1994;7:169-75. [ Links ]
10. Navarro H, Kolbach M, Repeto G, Guiraldes E, Harris P, Foradori A, Poggi H, Sanchez I. Correlation between phenotype and genotype in a group of patients with cystic fibrosis. Rev Med Chil 2002; 130:475-81. [ Links ]
11. Molina G, Gonzalez FJ, Cave R, Cornejo de M, Navarro S, Deglin M, Milinarsky A, Carvallo de P. Clinical and molecular genetic study of cystic fibrosis in the 5th region of Chile. Rev Med Chil 2002;130:850-8. [ Links ]
12. Gan KH, Veeze HJ, Van den Ouweland AM, Halley DJ, Scheffer H, Van der Hout A, Overbeek SE, De Jongste JC, Bakker W, Heijerman HG. A cystic fibrosis mutation associated with mild lung disease. N Engl J Med, 1995;333:95-99. [ Links ]
13. The Cystic Fibrosis Genotype-Phenotype Consortium. Correlation between genotype and phenotype in cystic fibrosis. N Engl J Med 1993;329:1308-13. [ Links ]
14. McKone E, Emerson S, Edwards K, Aitken M. Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet 2003;361:1671-6. [ Links ]
15. Accurso F, Sontag M. Seeking modifier genes in cystic fibrosis. Am J Respir Crit Care Med 2003;167:289-90. [ Links ]
16. Schwiebert EM, Egan ME, Hwang TH, Fulmer SB, Allen SS, Cutting GR, Guggino WB. CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell 1995;81:1063-1073. [ Links ]
17. Knowles M, Gatzy J, Boucher R. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Engl J Med 1981;305:1489-1495. [ Links ]
18. Davies JC, Geddes DM, Scallon M, Bush A, Jaffe A, Smith S, Davies MG, Alton EW. Confirmation of abnormal chloride ion secretion in the lower airway of children with cystic fibrosis. Pediatr Pulmonol 2000;20:293. [ Links ]
19. Quinton PM. The neglected ion: HCO3. Nat Med 2001;7:292-93. [ Links ]
20. Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM. Human beta-Defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 1997;88:553-560. [ Links ]
21. Yankaskas JR, Boucher RC. Ion transport properties of human fetal tracheal epithelial cells in primary cultures. Am Rev Respir Dis 1990;141:A163. [ Links ]
22. Pier GB, Grout M, Zaidi TS, Olsen JC, Johnson LG, Yankaskas JR, Goldberg JB. Role of mutant CFTR in hypersusceptibility of cystic fibrosis patients to lung infections. Science 1996;271:64-67. [ Links ]
23. Davies JC, Stern M, Dewar A, Caplen NJ, Munkonge FM, Pitt T, Sorgi F, Huang L, Bush A, Geddes DM, Alton EW. CFTR gene transfer reduces the binding of Pseudomonas aeruginosa to cystic fibrosis respiratory epithelium. Am J Respir Cell Mol Biol 1997;16:657-663. [ Links ]
24. Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med 1995;151:1075-1082. [ Links ]
25. Worlitzsch D, Tarran R, Ulrich M, et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest 2002;109:317-25. [ Links ]
26. Armstrong DS, Grimwood K, Carzino R, Carlin JB, Olinsky A, Phelan PD. Lower respiratory infection and inflammation in infants with newly diagnosed cystic fibrosis. BMJ 1995;310:1571-1572. [ Links ]
27. Konstan MW, Berger M. Current understanding of the inflammatory process in cystic fibrosis: onset and etiology. Pediatr Pulmonol 1997;24:137-142. [ Links ]
28. Tirouvanziam R, De Bentzmann S, Hubeau C, Hinnrasky J, Jacquot J, Peault B, Puchelle E. Inflammation and infection in naive human cystic fibrosis airway grafts. Am J Respir Cell Mol Biol 2000;23:121-127. [ Links ]
29. Weaver LT, Green MR, Nicholson K, Mills J, Heeley ME, Kuzemko JA, Austin S, Gregory GA, Dux AE, Davis JA. Prognosis in cystic fibrosis treated with continuous flucloxacillin from the neonatal period. Arch Dis Child, 1994;70:84-89. [ Links ]
30. Smyth A, Walters S. Prophylactic antibiotics for cystic fibrosis. Cochrane Database Syst Rev 2003;(3):1911. [ Links ]
31. McCaffery K, Olver RE, Franklin M, Mukhopadhyay S. Systematic review of antistaphylococcal antibiotic therapy in cystic fibrosis. Thorax 1999;54:380-383. [ Links ]
32. Smyth A,Walters S. Prophylactic antibiotics for cystic fibrosis. Cochrane Database Syst Rev 2003;(3):1912. [ Links ]
33. Ratjen F, Comes G, Paul K, Posselt HG, Wagner TO, Harms K. Effect of continuous antistaphylococcal therapy on the rate of P. aeruginosa acquisition in patients with cystic fibrosis. Pediatr Pulmonol 2001;31:13-6. [ Links ]
34. Stutman HR, Lieberman JM, Nussbaum E, Marks MI. Antibiotic prophylaxis in infants and young children with cystic fibrosis: a randomized controlled trial. J Pediatr 2002;140:299-305. [ Links ]
35. Valerius NH, Koch C, Hoiby N. Prevention of chronic Pseudomonas aeruginosa colonisation in cystic fibrosis by early treatment. Lancet 1991;338:725-726. [ Links ]
36. Doring G, Conway SP, Heijerman HG, Hodson ME, Hoiby N, Smyth A, Touw DJ. Antibiotic therapy against Pseudomona aeruginosa in cystic fibrosis: a European consensus. Eur Respir J 2000;16:749-67. [ Links ]
37. Szaff M, Hoiby N, Flensborg EW. Frequent antibiotic therapy improves survival of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection. Acta Paediatr Scand,1983;72:651-657. [ Links ]
38. Elborn JS, Prescott RJ, Stack BH, Goodchild MC, Bates J, Pantin C, Ali N, Shale DJ, Crane M. Elective versus symptomatic antibiotic treatment in cystic fibrosis patients with chronic Pseudomonas infection of the lungs. Thorax 2000;55:355-358. [ Links ]
39. Bren L, Aswani N. Elective versus symptomatic intravenous antibiotic therapy for cystic fibrosis. Cochrane Database Syst Rev 2001;(4): 2767. [ Links ]
40. Pegues DA, Carson LA, Tablan OC, FitzSimmons SC, Roman SB, Miller JM, Jarvis WR. Acquisition of Pseudomonas cepacia at summer camps for patients with cystic fibrosis. Summer Camp Study Group. J Pediatr 1994;124:694-702. [ Links ]
41. Muhdi K, Edenborough FP, Gumery L, OHickey S, Smith EG, Smith DL, Stableforth DE. Outcome for patients colonised with Burkholderia cepacia in a Birmingham adult cystic fibrosis clinic and the end of an epidemic. Thorax 1996;51:374-377. [ Links ]
42. Goss CH, Aitken ML, Otto K, Rubenfeld GD. Acquiring stenotrophomonas maltophilia does not reduce survival in patients with cystic fibrosis. Pediatr Pulmonol 2000;20:101-103. [ Links ]
43. OCarroll MR, Kidd TJ, Coulter C, Smith HV, Rose BR, Harbour C, Bell SC. Burkholderia pseudomallei: another emerging pathogen in cystic fibrosis. Thorax 2003;58:1087-1091. [ Links ]
44. Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, Vasiljev-K M, Borowitz D, Bowman CM, Marshall BC, Marshall S, Smith AL. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med 1999;340:23-30. [ Links ]
45. Le Brun PP, Brimicombe RW, Mannes GP, De Boer AH, Frijlink HW, Vinks AA, Heijerman HG. Advantages of colistin dry powder inhalation: Proof of principle. Pediatr Pulmonol 2000;20:285. [ Links ]
46. Auerbach HS, Williams M, Kirkpatrick JA, Colten HR. Alternate-day prednisone reduces morbidity and improves pulmonary function in cystic fibrosis. Lancet 1985;8457:686-688. [ Links ]
47. Rosenstein BJ, Eigen H. Risks of alternate-day prednisone in patients with cystic fibrosis. Pediatrics, 1991;87:245-246. [ Links ]
48. Balfour-Lynn I, Dezateux C. Corticosteroids and ibuprofen in cystic fibrosis. Thorax 1999;54:655. [ Links ]
49. Minicucci L, Severi G, cresta L, Giannattasio A, Lorini R, Haupt R. Impact of inhaled corticosteroids on the risk of early Pseudomonas aeruginosa acquisition in cystic fibrosis. Acta Paediatr 2003;92:684-87. [ Links ]
50. Konstan MW, Byard PJ, Hoppel CL, Davis PB. Effect of high-dose ibuprofen in patients with cystic fibrosis. N Engl J Med 1995;332:848-854. [ Links ]
51. Spencer H, Jaffé A. Newer therapies for cystic fibrosis. Current Paediatrics 2003;13:259-63. [ Links ]
52. Hoiby N. Diffuse panbronchiolitis and cystic fibrosis: East meets. Thorax, 1994;49:531-532. [ Links ]
53. Tredaniel J, Zalcman G, Gerber F, DAgay MF, Capron F, Frija J, Hirsch A. Diffuse panbronchiolitis: efficacy of low-dose erythromycin. Respir Med 1993;87:229-230. [ Links ]
54. Jaffe A, Francis J, Rosenthal M, Bush A. Long-term azithromycin may improve lung function in children with cystic fibrosis. Lancet 1998;351:420. [ Links ]
55. Wolter J, Seeney S, Bell S, Bowler S, Masel P, Mc Cormark J. Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: a randomised trial. Thorax. 2002;57:212-6. [ Links ]
56. Equi A, Balfour-Lynn IM, Bush A, Rosenthal M. Long term azithromycin in children with cystic fibrosis: a randomised, placebo-controlled crossover trial. Lancet 2002;360:978-84. [ Links ]
57. Phillips GE, Pike S, Jaffe A, Bush A. Active cyscle of breathing technique versus external high frequency oscillations in cystic fibrosis. Am J Respir Crit Care Med 1999;159:A687. [ Links ]
58. Van der Schans CP. Forced expiratory manoeuvres to increase transport of bronchial mucus: a mechanistic approach. Monaldi Arch Chest Dis 1997;52:367-370. [ Links ]
59. van der Schans, Prasad A, Main E. Chest physiotherapy compared to no chest physiotherapy for cystic fibrosis. Cochrane Database Syst Rev 2000;(2): 1401 [ Links ]
60. Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, Rosenstein BJ, Smith AL, Wohl ME. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. N Engl J Med 1994;331:637-642. [ Links ]
61. Shah P, Scott S, Fuchs H, Geddes D, Hodson M. Medium term treatment of stable stage cystic fibrosis with recombinant human DNase I. Thorax 1995;50:333-8. [ Links ]
62. Quan J, Tiddens H, Sy J, McKenzie S, Montgomery M, Robinson P, Wohl M, Konstan M; Pulmozyme Early Intervention Trial Study Group. A two-year randomized, placebo-controlled trial of dornase alfa in young patients with cystic fibrosis with mild lung function abnormalities J Pediatr 2001;139:813-20. [ Links ]
63. Suri R, Marshall L, Wallis C, Metcalfe C, Bush A, Shute J. Effects of recombinant human DNase and hypertonic saline on airway inflammation in children with cystic fibrosis. Am J Respir Crit Care Med 2002;166:352-5. [ Links ]
64. Dorin JR, Dickinson P, Alton EWFW, Smith SN, Geddes DM, Stevenson BJ, Kimber WL, Fleming S, Clarke AR, Hooper ML, Anderson L, Beddington RSP, Porteous DJ. Cystic fibrosis in the mouse by targeted insertional mutagenesis. Nature 1992;359:211-215. [ Links ]
65. Schidlow DV. Newer therapies for cystic fibrosis. Paediatr Respir Rev 2000;1:107-13. [ Links ]
66. Knowles MR, Hohneker KW, Zhou Z, Olsen JC, Noah TL, Hu P, Leigh MW, Engelhardt JF, Edwards LJ, Jones KR, Grossman M, Wilson JM, Johnson LG, Boucher RC. A controlled study of adenoviral-vector-mediated gene transfer in the nasal epithelium of patients with cystic fibrosis. N Engl J Med 1995;333:823-831. [ Links ]
67. Scott J, Higenbottam T, Hutter J, Hodson M, Stewart S, Penketh A, Wallwork J. Heart-lung transplantation for cystic fibrosis. Lancet 1988;2:192-194. [ Links ]
68. Aurora P, Wade A, Whitmore P, Whitehead B. A model for predicting life expectancy of children with cystic fibrosis. Eur Respir J 2000;16:1056-60. [ Links ]
69. Liou TG, Adler FR, Cahill BC, FitzSimmons SC, Huang D, Hibbs JR, Marshall BC. Survival effect of lung transplantation among patients with cystic fibrosis. JAMA 2001; 286:2683-9. [ Links ]
Correspondencia:
Dr. Luis Enrique Vega-Briceño
Lira 85 5to piso. Santiago de Chile
Casilla postal 114-D.
Laboratorio de Respiratorio Infantil
Teléfono: 0056(2)354-3767
Fax: 0056(2)247-3879


{kind=link}