










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Medica Herediana
Print version ISSN 1018-130XOn-line version ISSN 1729-214X
Rev Med Hered vol.18 no.2 Lima Apr./jun. 2007
Tratamiento quirúrgico de disección de aorta torácica ascendente en sindrome de marfán.
Surgical treatment of dissection of the ascending aorta in Marfan syndrome.
Montesinos Mosqueira Efraín1, Vasquez Kobashigawa Julio C2, 3, Rojas Peña Luis2, Peralta Rodriguez Julio2
1
Director, Programa de Cirugía de Tórax y Cardiovascular, Hospital Nacional Dos de Mayo, Lima, Perú.2
Cirujano, Programa de Cirugía de Tórax y Cardiovascular, Hospital Nacional Dos de Mayo, Lima, Perú3
Unidad de Epidemiología Clínica, Facultad de Medicina Alberto Hurtado. Universidad Peruana Cayetano Heredia. Lima, Perú.
RESUMEN
Se presentan 5 casos consecutivos de pacientes con Síndrome de Marfan y disección de aorta torácica ascendente, quienes fueron tratados quirúrgicamente con reemplazo de raíz aórtica (operación modificada de Bentall). Todos los pacientes tuvieron plena recuperación y al seguimiento post alta realizan actividades de la vida diaria sin problemas. (Rev Med Hered 2007;18:110-115).
PALABRAS CLAVE: Síndrome de Marfan, disección aórtica, operación de Bentall, aorta torácica ascendente.
SUMMARY
We present 5 consecutive patients with Marfan Syndrome and dissection of the ascending aorta that were surgically treated with aortic root replacement (modified Bentall operation). All patients had full recovery and returned to normal activities upon follow up. (Rev Med Hered 2007;18:110-115).
KEY WORDS: Marfan syndrome, aortic dissection, Bentall operation, ascending aorta.
INTRODUCCION
El Síndrome de Marfan es una enfermedad autosómica dominante del tejido conectivo que afecta principalmente el sistema músculo-esquelético, ocular y cardiovascular (1). Tiene una prevalencia de 2-3/10 000 habitantes y su pronóstico es principalmente determinado por la dilatación progresiva de la aorta torácica ascendente que se acompaña de dilatación del anillo valvular aórtico o anuloectasia de la válvula aórtica (2). Esta condición es frecuentemente complicada con disección aórtica cuando el diámetro del aneurisma es mayor de 5 cm, lo cual puede causar la muerte a temprana edad.
La disección aórtica es la condición letal más común de la aorta, y ocurre 3 veces más frecuentemente que la ruptura de un aneurisma abdominal aórtico. Los dos sistemas de clasificación más usados son el de DeBakey (tipos I, II, IIIa y IIIb) y Stanford (tipos A y B). El compromiso de la aorta torácica ascendente tiene el peor pronóstico y mayor mortalidad, correspondiendo al tipo I (aorta torácica ascendente, arco aórtico y aorta torácica descendente) y tipo II (sólo aorta ascendente) de la clasificación de DeBakey; y al tipo A de la clasificación de Stanford (3).
No hemos hallado referencias en la literatura nacional sobre disección de aorta torácica ascendente en Síndrome de Marfan. Presentamos aquí nuestra experiencia con el tratamiento quirúrgico de 5 pacientes consecutivos con esta condición.
RESULTADOS
El Programa de Cirugía de Tórax y Cardiovascular del Hospital Nacional Dos de Mayo (Lima, Perú) fue creado en febrero del año 1999. Desde entonces hasta agosto 2006, se operaron 5 pacientes con Síndrome de Marfan que presentaron disección aórtica tipo A de Stanford, asociado a aneurisma de aorta torácica ascendente e insuficiencia aórtica severa. Esto representa el 0,96 % de los casos de cirugía cardíaca con circulación extracorpórea operados en dicho período (n=517). La edad promedio fue 39,4 ± 14,25 años (rango: 27 a 62 años), siendo 3 pacientes del sexo femenino. La talla promedio fue de 1,75 ± 0,012 m (rango: 1,74 a 1,77cm). El tiempo desde el momento del diagnóstico definitivo de disección aórtica hasta el momento de la operación fue de 230 ± 280,7 días (rango, 14 a 720 días). Todos los pacientes tenían insuficiencia cardíaca congestiva grado III (clasificación de New York Heart Association) debido a insuficiencia aórtica severa. Además, presentaban dolor retroesternal y palpitaciones.
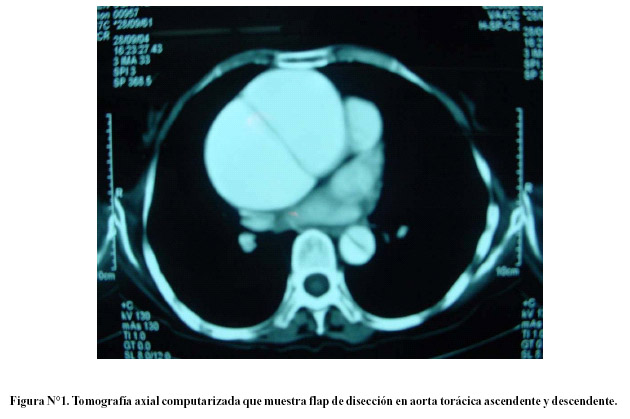
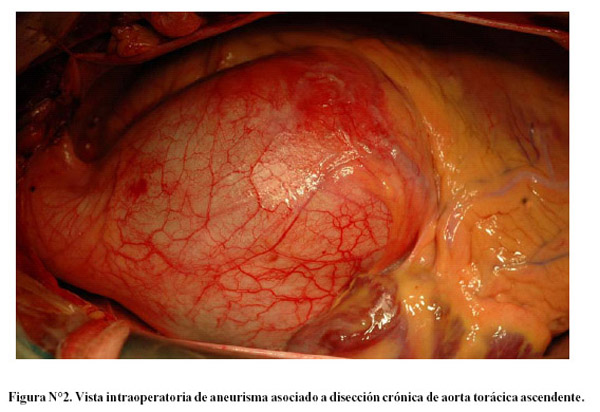
El diagnóstico definitivo se hizo con tomografía axial computarizada (Figura N°1) o ecocardiografía transesofágica. La operación realizada fue el reemplazo de raíz aórtica (válvula aórtica y aorta torácica ascendente) con injerto valvado (St. Jude Medical Aortic Valved Grafo; Model 23, 25 o 27 CAVG-404; St. Jude Medical Inc, Minnesota, USA) y reimplantación "en botón" de arterias coronarias (Figura N°2 y figura N°3).

En todos los casos se utilizó canulación arterial a través de arteria femoral común, y canulación venosa a través de aurícula derecha. Se administró cardioplegia retrógrada en seno coronario y cardioplegia anterógrada en las arterias coronarias. El injerto valvado fue cortado transversalmente obteniéndose dos segmentos que fueron anastomosados separadamente y luego unidos con sutura continua. Luego de inducir parada cardíaca, la secuencia seguida fue: aortotomía transversa, excisión de válvula aórtica, resección "en botón" de arterias coronarias, anastomosis distal de un segmento del injerto valvado a arco aórtico, implantación del otro segmento del injerto valvado conteniendo la válvula aórtica, reimplantación de botones coronarios y anastomosis entre los dos segmentos de injerto valvado. El tiempo de circulación extracorpórea fue de 255 ± 64,9 minutos (rango, 195 a 355 minutos) y el de clampado aórtico fue de 187 ± 39,4 minutos (rango, 140 a 228 minutos).
Adicionalmente, fue necesario realizar arresto circulatorio con hipotermia profunda, sin perfusión cerebral, en 3 pacientes (20, 35 y 40 minutos, respectivamente) con compromiso severo del arco aórtico que hacía imposible el uso de clamp aórtico. El tiempo de estadía postoperatoria en la unidad de cuidados intensivos fue de 5,6 ± 1,94 días (rango, 3 a 8 días), el tiempo de ventilación mecánica postoperatoria fue de 19,2 ± 16,6 horas (rango, 8 a 48 horas), y el tiempo total de hospitalización postoperatoria fue de 16,8 ± 3,96 días (rango, 13 a 22 días). Se utilizó por paciente 2,8 ± 2,48 unidades de paquetes globulares, 2,2 ± 1,48 unidades de plasma fresco congelado y 1,2 ± 0,83 unidades (aféresis) de plaquetas. El tiempo de circulación extracorpórea más prolongado (355 minutos) ocurrió en una paciente que requirió revisión de anastomosis entre los dos segmentos del injerto valvado debido a que el ángulo entre ellos era muy pronunciado y ocasionaba estenosis relativa del lumen.
Dos pacientes tuvieron complicaciones postoperatorias: Una paciente requirió reesternotomía a las 6 horas postoperatorias para control de sangrado mediastinal asociado a coagulopatía, para lo cual se utilizó equipo de autotransfusión disminuyéndose la necesidad de transfusión sanguínea. Otra paciente tuvo dehiscencia superficial del sitio de canulación de arteria femoral, la cual fue suturada dos semanas luego de la operación. Todos los pacientes tuvieron un ecocardiograma 1-3 meses luego de la operación, que mostró válvula mecánica normofuncionante con flujo normal en injerto tubular. El tiempo de seguimiento promedio fue de 18,6 ±11,19 meses (rango: 1 a 30 meses). La misma paciente que requirió reesternotomía para control de sangrado postoperatorio desarrolló bacteremia a Staphylococcus aureus, 9 meses luego de la operación. A pesar de que no se halló vegetaciones en ecocardiografía transesofágica, se administró antibióticos endovenosos por 8 semanas por posible endocarditis infecciosa, siendo su recuperación completa. Dos pacientes tienen disección crónica en toda la aorta remanente, y requieren TAC anuales para vigilar probable dilatación progresiva de aorta torácica y abdominal.
A la fecha, todos los pacientes se encuentran laboralmente activos y desarrollando actividades normales de la vida diaria, con controles periódicos de anticoagulación (INR: 2 -3) para prevenir trombosis de la prótesis valvular aórtica.
DISCUSION
La disección aórtica demanda inmediata respuesta terapéutica. Sin tratamiento, aproximadamente 50% de los pacientes con disección aguda de aorta torácica ascendente mueren dentro de las primeras 48 horas (3), por ello se recomienda su tratamiento quirúrgico inmediato. En nuestros pacientes, el tiempo promedio de enfermedad fue de 266 días (rango 14 a 720 días), lo cual los convierte en casos de disección aórtica subaguda o crónica.
El tratamiento quirúrgico de disección aórtica tipo A de Stanford es técnicamente complejo y se hace a través de una esternotomía mediana bajo anestesia general. Requiere uso de circulación extracorpórea, existiendo varias opciones para canulación arterial como la arteria subclavia derecha, que permite realizar perfusión aórtica anterógrada, siempre y cuando se acceda al lumen verdadero. Debido a la dificultad técnica que esto implica, la mayoría de cirujanos prefiere la canulación de una de las arterias femorales para realizar perfusión arterial retrógrada.
Durante la operación, se induce arresto cardíaco con infusión de cardioplegia retrógrada en seno coronario, y también se administra cardioplegia anterógrada directamente a las arterias coronarias. Con frecuencia, también es necesario hacer arresto circulatorio con hipotermia profunda (14 oC), para lo cual se drena prácticamente toda la sangre del paciente a un colector externo de la "máquina corazón-pulmón", obteniéndose un campo operatorio exsanguine. Esta última técnica es tolerada por el cerebro hasta por 30 minutos sin consecuencias negativas; pero, períodos más prolongados requieren métodos de protección cerebral para disminuir la probabilidad de daño cerebral. Ninguno de nuestros pacientes tuvo secuela neurológica central, a pesar de que dos de ellos tuvieron más de 30 minutos de arresto circulatorio.
En el año 1972, se estimaba que el promedio de vida de las personas con Síndrome de Marfan era de 37 años, siendo la mayoría de muertes prematuras debido a disección de aorta ascendente (4). Esta condición puede causar la muerte por uno o más mecanismos: hemorragia masiva en mediastino o hacia cavidad pleural, taponamiento cardíaco por hemopericardio masivo, falla ventricular izquierda por insuficiencia aórtica aguda, infarto de miocardio por oclusión del origen de las arterias coronarias, o accidente cerebrovascular isquémico por oclusión de lumen verdadero de las arterias carótidas como extensión de la disección desde el arco aórtico (5).
El procedimiento estándar para disección aórtica tipo A de Stanford en pacientes con Síndrome de Marfan es la operación de Bentall modificada. Esta operación reemplaza la raíz aórtica y utiliza un injerto tubular valvado (tubo de Dacron unido a válvula mecánica bivalva) al cual el cirujano conecta las arterias coronarias. De este modo se corrige la anuloectasia aórtica y la insuficiencia valvular aórtica asociada, y también se reemplaza la aorta dañada por la disección. En contraste con otros pacientes con aneurisma y/o disección de aorta ascendente, no es recomendable reparar (resuspender) y preservar la válvula aórtica nativa en pacientes con Síndrome de Marfan porque a largo plazo el anillo valvular tiende a dilatarse y la válvula nuevamente se vuelve incompetente.
Series recientes sobre tratamiento quirúrgico de disección aórtica aguda tipo A (Stanford) muestran una mortalidad operatoria de 2,4 a 2,8% y una mortalidad hospitalaria postoperatoria de 17,6% a 31% (6,7,8). En estas series, aproximadamente 11% de pacientes tienen Síndrome de Marfan (8). Una de las pocas series reportadas de disección tipo A crónica (9) incluyó 77 pacientes operados en el Hospital de la Pitie Salpetriere (Paris, Francia) en el período 1981-2001. Estos pacientes habían sobrevivido el episodio de disección aguda sin tratamiento quirúrgico, y luego la mayoría desarrolló insuficiencia valvular aórtica (n=60). Aunque se usaron diferentes técnicas quirúrgicas dependiendo de los hallazgos, el único factor de riesgo para mortalidad que se identificó fue extensión de la disección. Esta serie tuvo una frecuencia de accidente cerebrovascular de 2,5%, una mortalidad hospitalaria de 10% y una sobrevida de 42% a los 12 años.
Los resultados son aún mejores cuando se realizan operaciones electivas en pacientes con Síndrome de Marfan para el tratamiento de aneurisma de aorta ascendente no complicado, sin disección aórtica. Aquí, la mortalidad hospitalaria postoperatoria es de 3,3%, según la experiencia acumulada de 10 centros de referencia a nivel mundial que en conjunto operaron 675 pacientes entre 1968-1996 (10). La mayoría de cirujanos recomienda tratamiento quirúrgico electivo cuando la aorta torácica ascendente alcanza un diámetro de 5 cm, para evitar mortalidad por ruptura o situaciones de alto riesgo como disección aguda (11).
Las innumerables dificultades para realizar operaciones cardíacas en los hospitales del Ministerio de Salud de nuestro país y el alto costo de los insumos necesarios ayudan a explicar por qué nuestros pacientes recibieron tratamiento quirúrgico tardío luego del episodio agudo de disección. A pesar de ello, los resultados obtenidos son comparables a los de la literatura mundial y estos pacientes han podido retornar a una vida productiva.
REFERENCIAS BIBLIOGRAFICAS
1. Nollen G, Mulder B. What is new in the Marfan syndrome?. Int J Cardiol 2004; 97:103-8. [ Links ]
2. Kazui T, Yamashita K, Terada H, et al. Later reoperation for proximal aortic and arch complications after previous composite graft replacement in Marfan patients. Ann Thorac Surg 2003; 76:1203-8. [ Links ]
3. Green R, Kron IL. Aortic dissection. En: Cohn LH, Edmunds Jr. LH, editors. Cardiac Surgery in the adult. 2nd edition. Mc-Graw Hill; 2003. p. 1095-1122. [ Links ]
4. Van Karnebeek CDM, Naeff MSJ, Mulder BJM, Hennekam RCM, Offringa M. Natural history of cardiovascular manifestations in Marfan syndrome. Arch Dis Child 2001; 84:129-37. [ Links ]
5. Treasure T. Cardiovascular surgery for Marfan syndrome. Heart 2000; 84:674-8. [ Links ]
6. Januzzi J, Isselbacher E, Fattori R, et al. Characterizing the young patient with aortic dissection: results from the international registry of aortic dissection (IRAD). J Am Coll Cardiol 2004; 43:665-9. [ Links ]
7. Chiappini B, Tan EM, Morshuis W, Kelder H, Dossche K, Schepens M. Surgery for acute type A aortic dissection: is advanced age a contraindication?. Ann Thorac Surg 2004; 78:585-90. [ Links ]
8. Kallenbach K, Oelze T, Salcher R, et al. Evolving strategies for treatment of acute aortic dissection type A. Circulation 2004;110: 243 –249. [ Links ]
9. Jault F, Rama A, Lievre L, et al. Chronic dissection of the ascending aorta: surgical results during a 20-year period (previous surgery excluded). Eur J Cardiothorac Surg 2006; 29(6):1041-5. [ Links ]
10.Gott VL, Greene PS, Alejo DE, et al. Replacement of the aortic root in patients with Marfan´s syndrome. N Engl J Med 1999; 340:1307-13. [ Links ]
11.Braverman A. Timing of aortic surgery in the Marfan syndrome. Curr Opin Cardiol 2004; 549-50. [ Links ]
Correspondencia:
Dr. Efraín Montesinos Mosqueira
Programa de Cirugía de Tórax y Cardiovascular
Hospital Nacional Dos de Mayo
Av Parque de la Medicina s/n,
Lima Peru.

