










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Medica Herediana
versión impresa ISSN 1018-130Xversión On-line ISSN 1729-214X
Rev Med Hered v.18 n.3 Lima jul.-set. 2007
Apoplejía pituitaria: Reporte de dos casos.
Pituitary apoplexy: Two cases report.
Pinto Sánchez Juan Fernando1, Villena Chavez Jaime2, Villena Pacheco Arturo2, Seclén Santisteban Segundo2, Ferrufino Llach Juan Carlos3, Pinto Valdivia Miguel2, Huachín Soto Miluska1, Jasso Huamán Luís1, Guevara Linares Ximena1.
1
Médico Residente de Endocrinología. Hospital Nacional Cayetano Heredia. Facultad de Medicina Alberto Hurtado. Universidad Peruana Cayetano Heredia. Lima, Perú.2
Médico Asistente del Servicio de Endocrinología. Hospital Nacional Cayetano Heredia. Profesor del Departamento de Medicina. Facultad de Medicina Alberto Hurtado. Universidad Peruana Cayetano Heredia. Lima, Perú.3
Médico Asistente del Servicio de Patología. Hospital Nacional Cayetano Heredia. Profesor Principal del Departamento de Patología. Facultad de Medicina Alberto Hurtado. Universidad Peruana Cayetano Heredia. Lima, Perú.
RESUMEN
Se presentan dos pacientes del Hospital Nacional Cayetano Heredia, el primero con un adenoma pituitario no funcionante sin diagnóstico previo, el segundo con acromegalia; inicialmente tuvieron los diagnósticos de hemorragia subaracnoidea y meningitis aguda respectivamente, y durante la evolución fueron diagnosticados de apoplejía pituitaria. Se describe la evolución clínica de ambos casos, y se hace una revisión bibliográfica del tema. Se plantea la necesidad de considerar el cuadro de apoplejía pituitaria dentro del diagnóstico diferencial cuando existe cefalea, oftalmoplejía y compromiso visual. (Rev Med Hered 2007;18:165-172).
PALABRAS CLAVE: Apoplejía pituitaria, acromegalia, adenoma hipofisiario.
SUMMARY
We report two cases from Hospital Nacional Cayetano Heredia, the first with unknown non-functioning pituitary adenoma, the second one with acromegaly, whose initial diagnosis were subarachnoid haemorrhage and acute meningitis respectively. A classic picture of pituitary apoplexy was developed in the evolution. We describe the clinical progress in both cases and make a review of the medical literature. Pituitary apoplexy must be considered in the differential diagnosis when headache, ocular paresis, and visual impairment are present.(Rev Med Hered 2007;18:165-172).
KEYWORDS: Pituitary apoplexy, acromegaly, pituitary adenoma.
INTRODUCCIÓN
La apoplejía pituitaria, una emergencia endocrina, es una condición rara, cuyo diagnóstico generalmente es confundido con entidades como hemorragia subaracnoidea o meningitis aguda, por lo que es necesario incluir apoplejía pituitaria en el diagnóstico diferencial cuando se presentan cefalea, anormalidades del campo visual, oftalmoplejía, y alteración del nivel de conciencia (1). En el Hospital Nacional Cayetano Heredia se presentaron dos casos, cuyos diagnósticos iniciales fueron hemorragia subaracnoidea y meningitis aguda respectivamente, diagnosticándose posteriormente apoplejía pituitaria. La importancia en establecer un diagnóstico y tratamiento precoz en esta patología estimuló a la realización de la presente publicación, en la que se reportan los casos clínicos y se revisa la literatura.
Caso clínico 1
Varón de 48 años, carpintero, con cefalea esporádica de larga evolución, negando otro antecedente relevante. Acude a emergencia con 6 horas de enfermedad, refiriendo cefalea holocránea, pulsátil-opresiva, rápidamente progresiva de gran intensidad (10/10), sin respuesta a analgésicos orales, asociada a náuseas y vómitos no explosivos.
Al ingreso tuvo una presión arterial de 120/80 mmHg, frecuencia cardiaca de 86 latidos por minuto, temperatura oral de 37.8 °C. Durante el examen el paciente se encontraba somnoliento, con hemianopsia bitemporal y rigidez de nuca. No tenía papiledema.
La tomografía cerebral sin contraste no mostró alteraciones; la punción lumbar tuvo una presión de apertura de 37 cm de agua, el estudio del líquido céfalo-raquídeo fue compatible con hemorragia subaracnoidea. Se inició tratamiento con nimodipino. El paciente permaneció somnoliento y subfebril, a las 48 horas presentó diplopía por parálisis del VI par craneal derecho, el séptimo día presentó ceguera en el ojo derecho asociada a parálisis del IV par craneal, con imposibilidad de dirigir la mirada hacia abajo.
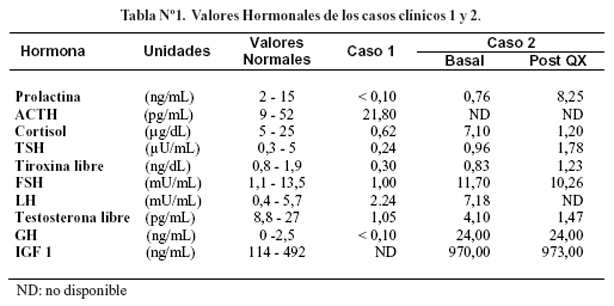
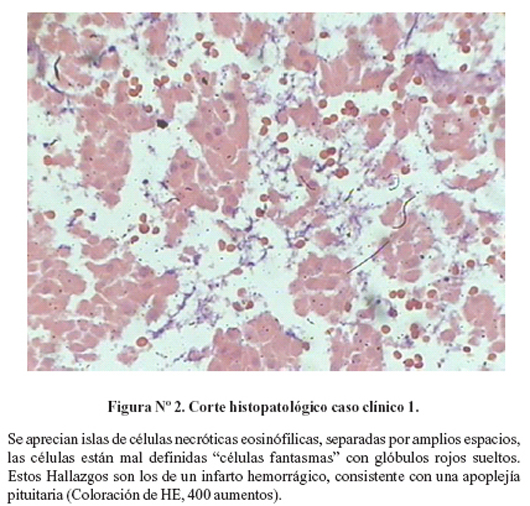
Se realizó una panangiografía cerebral, que no mostró alteraciones. La resonancia magnética cerebral mostró una tumoración hipofisiaria con extensión supraselar y hemorragia central (Figura Nº1). La evaluación del perfil hormonal mostró panhipopituitarismo (Tabla Nº1). Se inició tratamiento con hidrocortisona 100 mg EV cada 8 horas. A los 14 días de ingreso se sometió a cirugía de resección transesfenoidal. La evaluación histopatológica confirmó la presencia de apoplejía pituitaria (Figura Nº2).
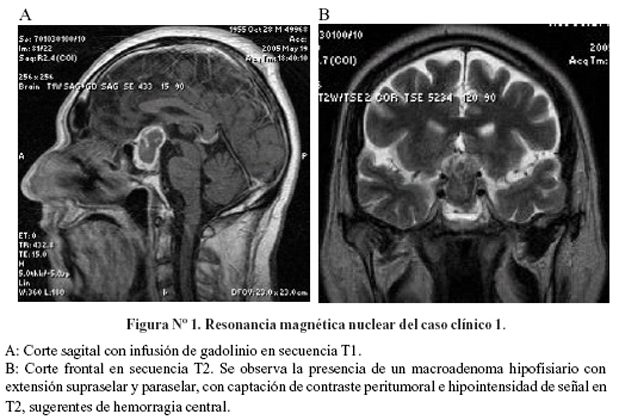
A partir del 2do día postoperatorio mostró recuperación del campo visual y motilidad ocular. Se continuó terapia de reemplazo hormonal con prednisona, levotiroxina y testosterona. El control tomográfico post quirúrgico reveló restos tumorales supraselares, por lo que se administró radioterapia con 50,4 Gy por acelerador lineal en 28 sesiones.
Caso clínico 2
Varón de 30 años de edad, con diagnóstico de otitis media aguda de 3 días de evolución, con tratamiento antibiótico y analgésico por vía oral. Refería dos días de cefalea opresiva, retro-orbitaria, de gran intensidad (8/10). Diez horas antes de su ingreso presentó escalofríos, sudoración profusa y vómitos explosivos.
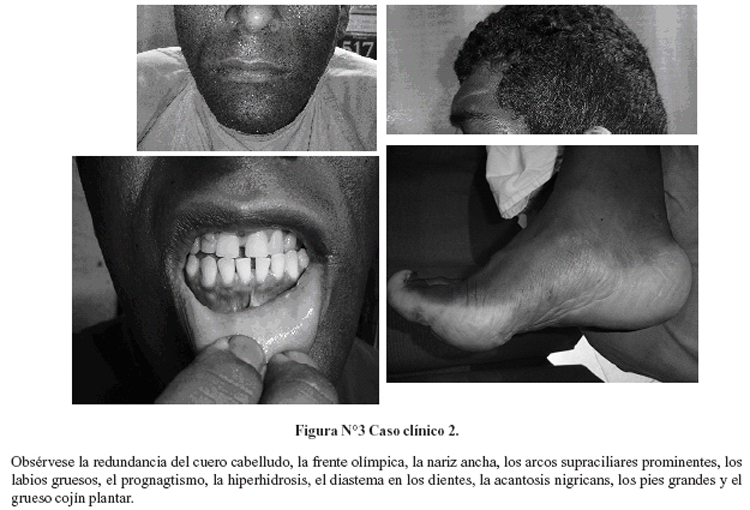
Al ingreso, presión arterial de 150/100 mmHg, frecuencia cardiaca de 72 latidos por minuto y temperatura oral de 39,4 °C, con índice de masa corporal de 34 Kg/m2 y perímetro de cintura de 106 cm. Somnoliento, con fascies acromegálica (Figura Nº3), hiperhidrosis, vello corporal abundante, acantosis nigricans y voz ronca. La otoscopía mostró perforación timpánica bilateral, el fondo de ojo fué normal, se evidenció parálisis del VI par craneal izquierdo.
A las 3 horas de evolución presentó hemianopsia bitemporal a predominio izquierdo. La tomografía cerebral mostró un macroadenoma hipofisiario heterogéneo. Se realizó una punción lumbar, con presión de apertura de 5 cm. de agua. El estudio del líquido céfalo raquídeo fue normal. La resonancia magnética nuclear cerebral mostró un macroadenoma hipofisiario supra y paraselar, con necrosis central (Figura Nº 4).
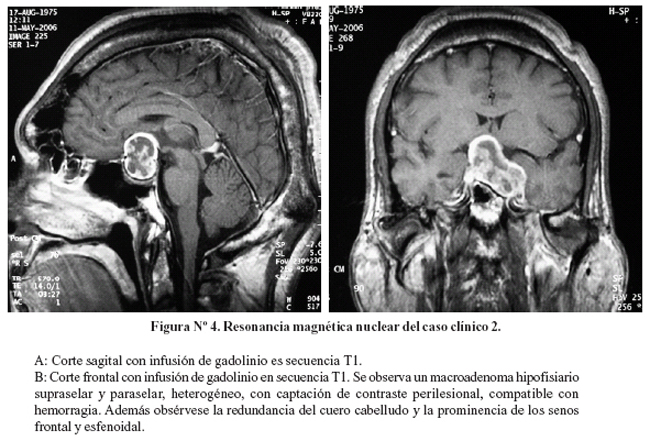
La evaluación serológica confirmó el diagnóstico de acromegalia, además mostró prolactina suprimida, hipocortisolismo, hipogonadismo, e hiperglicemia (Tabla Nº1).
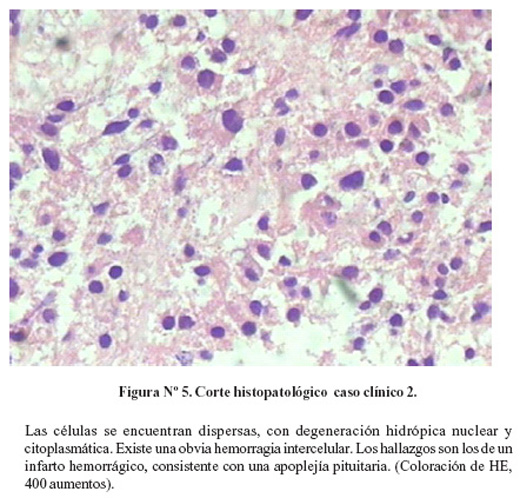
Se inició tratamiento con hidrocortisona 100 mg. EV cada 8 horas, insulina regular en infusión y antihipertensivos. Después de 30 días de hospitalización se realizó cirugía transcraneal izquierda. El estudio histopatológica confirmó el diagnóstico de apoplejía pituitaria (Figura Nº5). La parálisis del VI par craneal se resolvió a las 24 hrs. de la cirugía, sin embargo permaneció con hemianopsia temporal izquierda leve. Los valores postquirúrgicos de IGF-1 permanecieron elevados. La hipertensión arterial e hiperglicemia se resolvieron rápidamente. Recibió reemplazo hormonal con 7,5 mg de prednisona, quedando pendiente la radioterapia.
DISCUSIÓN
La apoplejía pituitaria es un síndrome compuesto por cefalea, náuseas, alteración visual, oftalmoplejía y trastorno del sensorio; siendo consecuencia de hemorragia o infarto en un adenoma hipofisiario (1).
Descrita inicialmente en 1898 por Pearce Bailey en un acromegálico con cefalea, náuseas, vómitos, fiebre, oftalmoplejía y pérdida visual, fue denominada apoplejía pituitaria por primera vez por Brougman en 1950 con el reporte de 5 casos (1).
La incidencia de la apoplejía pituitaria es variable. En estudios anatomopatológicos de adenomas hipofisiarios se describe una frecuencia de 1 a 20% (1), presentando el 0,6 a 9% el cuadro clínico clásico de apoplejía pituitaria, el resto son presentaciones subclínicas (2-4).
Es más frecuente en varones. Se presenta desde los 6 hasta los 88 años, siendo más frecuentes entre los 46 y 56 años.
En el 81-87%, la apoplejía pituitaria es la primera manifestación del adenoma hipofisiario. Entre 58-80% de las adenomas son no funcionantes (1-6). Ambos casos presentados correspondieron a varones, en el primer caso fue la primera manifestación del adenoma, en el segundo se presentó en un caso de acromegalia.
Los adenomas, irrigados por la arteria hipofisiaria inferior (1), presentan vasculopatía por endarteritis, que los hace propensos a hemorragia (2). La hemorragia produce edema tisular con incremento abrupto de la presión intraselar, compresión del sistema porta (que irriga a la hipófisis anterior), e isquemia hipofisiaria. La distensión meníngea y la compresión de las estructuras adyacentes desencadenan el cuadro clínico clásico (7). La presión intraselar en casos de apoplejía pituitaria es extremadamente alta, con valores medios de 47 mmHg (8).
La presentación clínica es rápida, con expresión total en horas a 2 días (2,5), en ambos casos presentados el tiempo de presentación fue menor de 10 horas. La triada clásica es cefalea, oftalmoplejía y alteración en el nivel de conciencia (3), presente en ambos casos reportados. La cefalea es el síntoma más frecuente, presente en el 84%, siendo intensa, súbita, unilateral y retroorbitaria (2,5), causada por distensión, irritación meníngea, y compresión del trigémino. En el 24% se acompaña de nauseas y vómitos (2).
La compresión del quiasma óptico produce alteración del campo y la agudeza visual hasta en el 66% de los casos (2). Inicialmente se produce cuadrantopsia bitemporal superior, que puede progresar a ceguera total (1).
La oftalmoplejía es más frecuente que la afección del campo visual (1,2), la compresión del seno cavernoso altera los nervios oculomotores en el 43-69% (2-6), siendo el III par el más susceptible, la afección del IV par es infrecuente (1,2,9). La compresión de la arteria carótida interna puede provocar alteración en el nivel de conciencia, produciendo coma hasta en el 3% (2,9); también puede producir un soplo retroorbitario por turbulencia sanguínea (10). La compresión del sifón carotídeo provoca disfunción hemisférica, convulsiones y hemiplejía (2,9). La compresión simpática puede desencadenar un síndrome de Horner (1).
La compresión hipotalámica puede provocar diabetes insípida, hipotensión, alteración en la termorregulación y arritmias cardiacas (1). Se presenta fiebre hasta en el 24% (3).
La irritación meníngea y liberación de sustancias vasoactivas desde el adenoma y el hipotálamo puede producir vasoespasmo, con déficit neurológico focal transitorio (1,11).
La hemorragia subaracnoidea con apoplejía pituitaria es una condición rara pero posible (5,12), puesto que los adenomas pituitarios con frecuencia se asocian a aneurismas (13).
La apoplejía pituitaria inicialmente es confundida con hemorragia subaracnoidea en el 65% de los casos (2), con meningitis en el 29% (2), con migraña y con accidente cerebrovascular (3), además se debe considerar dentro del diagnóstico diferencial de neuritis óptica, encefalitis, sinusitis, trombosis del seno cavernoso, absceso pituitario (2) y en metástasis a hipófisis, siendo las más frecuentes las neoplasias de pulmón y mama (14). En el primer caso reportado, el diagnóstico inicial fue hemorragia subaracnoidea, sospechándose la presencia de un aneurisma en el sifón carotídeo, por lo que se requirió la realización de una panangiografía cerebral. En el segundo caso se sospechó el cuadro de meningitis bacteriana con punto de partida ótico. En ambos casos se descartó estas entidades, estableciéndose el diagnóstico de apoplejía pituiaria.
La mayoría de veces no se identifican factores precipitantes (2), el primer caso reportado no tuvo un desencadenante evidente, existiendo factores de riesgo en el 30-40% (3,15), siendo la hipertensión arterial el factor más importante (1-6), presente en el segundo caso. Los factores de riesgo son: 1. Disminución del flujo sanguíneo por incremento transitorio en la presión intracraneal (tos, ventilación mecánica con presión positiva, trauma, angiografía, neumoencefalografía, mielografía, punción lumbar) y fluctuaciones en la presión arterial (hipotensión durante cirugía cardiaca, laminectomía y hemodiálisis); 2. Alteraciones microvasculares (diabetes mellitus, hipertensión arterial, y radioterapia); 3. Estimulación glandular en el embarazo, pruebas dinámicas con GnRH y TRH, cirugías y enfermedades severas (sepsis, infarto cardiaco, por incremento de ACTH) y 4. Anticoagulación (15).
La tomografía axial computarizada supera a la resonancia magnética para detectar hemorragia aguda (1), sin embargo es poco sensible para detectar masas hipofisiarias (2,3,13).
La resonancia magnética es diagnóstica en el 91% de lesiones pituitarias, con una sensibilidad cercana al 100% (1-3). Las secuencias T2 son útiles para el diagnóstico de sangrado agudo, observándose señales hipointensas. En hemorragia subaguda, las secuencias T1 muestran áreas hiperintensas. La hemorragia crónica muestra áreas hipointensas (2,3,6,9). En caso de infarto, la captación de contraste peritumoral es menor, la secuencia de difusión muestra hiperintensidad, y el coeficiente de difusión aparente es restrictivo (16).
La sospecha de un aneurisma carotídeo requiere de una angiografía carotídea, siendo requerida en el 17-20% de los casos (3,5). La angioresonancia es una alternativa (6).
Existe disfunción central cuando los valores de hormonas periféricas están disminuidos y su contraparte central es "inadecuadamente normal"; por haber falla en la regulación (9). En el 73-81% existe alteración hormonal (2,3), en el 76-79% hay insuficiencia gonadotropa (3,5), en el 60-76% del corticotropo (3,5), y en el 50-57% del tirotropo (3,5). Diabetes insípida está presente hasta en el 8% (2). Los valores de prolactina son pronósticos, siendo los valores mayores de 3,5 ng/ml indicadores de viabilidad celular, con posibilidad de recuperación (8).
El manejo implica monitoreo hemodinámico, hidroelectrolítico y administración de corticosteroides a dosis altas (1-6), superando la posible insuficiencia adrenal, mejorando los defectos en el campo visual y la oftalmoplejía (1,2). Se recomienda hidrocortisona, 100 mg. EV cada 6-8 horas, debiendo disminuirse la dosis en 48 a 72 horas (13).
El tratamiento quirúrgico se recomienda en la mayoría de pacientes (1-6). La cirugía antes de los 7 días mejora el pronóstico visual (1,3), ya que el nervio óptico resiste a la isquemia, si es descomprimido antes de la semana (13), el tiempo quirúrgico no influye sobre la oftalmoplejía (3,5). En ambos casos presentados la oftalmoplejía se resolvió postquirúrgicamente, permaneciendo el deterioro del campo visual en el segundo caso. La cirugía es urgente cuando existe deterioro del nivel de conciencia o déficit visual (2-6).
Los hallazgos quirúrgicos consisten en tensión de la dura madre y hemorragia, siendo esta aguda en el 8% (2). Histológicamente se evidencia hemorragia, necrosis, células fantasmas y células pituitarias edematosas (3,5).
Cuando no existe alteración en el nivel de conciencia ni déficit visual pude optarse por tratamiento conservador, que consiste en corticoterapia y manejo hidroelectrolítico (13,17), la oftalmoplejía mejora, sin embargo, la posibilidad de recurrencia del tumor es mayor (2,13), al igual que la probabilidad de hipopituitarismo (13).
La radioterapia se aplica en el 74% de los casos de manera temprana (2). Está indicada cuando la resección quirúrgica es incompleta o cuando existe recurrencia tumoral (5).
La mortalidad en apoplejía pituitaria llega al 2%, la oftalmoplejía mejora en la casi totalidad de pacientes, el campo visual en el 95%, y la agudeza visual en el 88% (2).
Concluimos señalando que esta emergencia endocrinológica tiene un diagnóstico laborioso, como lo han mostrado ambos casos presentados, siendo fundamental la sospecha clínica, y la confirmación diagnóstica mediante resonancia magnética cerebral. Es necesario incluir a apoplejía pituitaria en el diagnóstico diferencial cuando se presentan cefalea, anormalidades del campo visual, oftalmoplejía, y alteración del nivel de conciencia. La corticoterapia a dosis elevadas siempre es necesaria, pudiendo individualizarse la necesidad quirúrgica de acuerdo al compromiso visual y alteración de conciencia. Brindando el tratamiento oportuno y correcto el pronóstico a largo plazo es bueno.
En ambos casos presentados los pacientes permanecen con capacidad para desarrollar una vida normal, sin limitaciones importantes para las actividades de la vida diaria.
REFERENCIAS BIBLIOGRAFICAS
1. Liu J, Rovit R, Couldwell W. Pituitary apoplexy. Semin Neurosurg 2001; 12(3): 315- 320. [ Links ]
2. Semple P, Webb M, De Villiers J, Laws E. Pituitary apoplexy. Neurosurgery 2005; 56(1): 65-73. [ Links ]
3. Sibalt L, Ball S, Connolly V, et al. Pituitary apoplexy: A review of clinical presentation, management and outcome in 45 Cases. Pituitary 2004; 7(3): 157-163. [ Links ]
4. Ayuk J, Mc Gregor E, Mitchell R, Gittoes N. Acute Management of pituitary apoplexy – surgery or conservative management?. Clin Endocrinol 2004; 61(6): 747-752. [ Links ]
5. Randeva H, Schoebel J, Byrne J, Esiri M, Adams C, Wass J. Classical pituitary apoplexy: clinical features, management and outcome. Clin Endocrinol 1999; 51(2): 181-188. [ Links ]
6. Dubuisson A, Beckers A, Stevenaert A. Classical pituitary tumor apoplexy: clinical features, management and outcomes in a series of 24 patients. Clin Neurol Neurosurg 2007; 109(7): 63-70. [ Links ]
7. Arafah B, Prunty D, Ybarra J, Hlavin M, Selman W. The Dominant Role of Increased Intrasellar Pressure in the Pathogenesis of Hypopituitarism, Hyperprolactinemia and Headaches in Patients with Pituitary Adenomas. J Clin Endocrinol Metab 2000; 85(5): 1789-1793. [ Links ]
8. Zayour D, Selman W, Arafah B. Extreme elevation of intrasellar pressure in patients with pituitary tumor apoplexy: Relation to pituitary function. J Clin Endocrinol Metab 2004; 89(11): 5649-5654. [ Links ]
9. Lee C, Cho A, Carter W. Emergency Department presentation of pituitary aplopexy. Am J Emerg Med 2000; 18(3): 328-331. [ Links ]
10.Watson S, Francis I, Coroneo M. Pituitary apoplexy presenting with an orbital bruit. Clin Experim Ophthalm 2002; 30(4): 305-307. [ Links ]
11.Pozzati E, Frank G, Nasi MT,Giuliani G. Pituitary apoplexy, bilateral carotid vasospasm, and cerebral infarction in a 15 year old boy. Neurosurgery 1987; 20(1): 56-59. [ Links ]
12.Laidlaw J, Tress B, Gonzales M, Wray A, Hoe W, OBrien J. Coexistence of aneurismal subarachnoid haemorrhage and pituitary apoplexy: Case report and review of the literature: J Clin Neuroscien 2003; 10(4): 478-482. [ Links ]
13.Maccagnan P, Macedo C, Kayath M, et al. Conservative Management of Pituitary Apoplexy: A Prospective Study. J Clin Endocrinol Metab 2004; 80(7): 2190-2197. [ Links ]
14.Hanna F, Williams O, Davies J, Dawson T, Neal J, Scanlon M. Pituitary apoplexy following metastasis of bronchogenic adenocarcinoma to a prolactinoma – Case Report. Clin Endocrinol 1999; 51(3): 377-381. [ Links ]
15.Biousse V, Newman N, Oyesiku N. Precipitating factors in pituitary apoplexy. J Neurol Neurosurg Psychiatry 2001; 71(4): 542 – 545. [ Links ]
16.Rogg J, Tung G, Anderson G, Cortez S. Pituitary apoplexy: Early detection with diffusion-Weighted MR imaging. Am J Neuroradiol 2002; 23(7): 1240-1245. [ Links ]
17.Agrawal D, Kumar A. Visual outcome of blind eyes in pituitary apoplexy after transsphenoidal surgery: a series of 14 eyes. Surg Neurol 2005; 63(1): 42-46. [ Links ]
Correspondencia:
Dr. Juan Fernando Pinto Sánchez
Servicio de Endocrinología
Hospital Nacional Cayetano Heredia
Av. Honorio Delgado s/n San Martín de Porres
Lima 31, Perú
Correo Electrónico: jfpinto@hotmail.com

