










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Medica Herediana
Print version ISSN 1018-130XOn-line version ISSN 1729-214X
Rev Med Hered vol.19 no.1 Lima Jan./mar 2008
Conversatorio Clínico Patológico en el Hospital Nacional Arzobispo Loayza_2008_01.
Clinical Case at the Hospital Nacional Arzobispo Loayza_2008-1.
Editor : Dr. Enrique Cipriani 1
Discusión clínica : Dr. Hugo Umeres 2
Discusión patológica : Dr. César Chian 3
Residente : Dr. Germán Ramos 4, Dr. Edison Sedano5
1
Profesor Principal del Departamento de Medicina. Universidad Peruana Cayetano Heredia. Lima, Perú.2
Profesor Auxiliar del Departamento de Medicina. Universidad Peruana Cayetano Heredia. Lima, Perú.3
Profesor Auxiliar del Departamento de Patología. Universidad Peruana Cayetano Heredia. Lima, Perú.4
Médico residente de tercer año de Medicina. Universidad Peruana Cayetano Heredia. Lima, Perú.5
Médico residente de primer año de Medicina. Universidad Peruana Cayetano Heredia. Lima, Perú.
Caso Clínico
Paciente mujer de 38 años, natural de Ayacucho, procedente de Lima. Desde aproximadamente un año antes del ingreso, nota parestesias y disestesias en miembro inferior derecho. Diez meses antes del ingreso se agregan debilidad en pierna derecha y parestesias en brazo derecho y pierna izquierda. Ocho meses antes del ingreso presenta vómitos de contenido bilioso que se autolimitan y es hospitalizada durante un mes, evidenciándose disminución de la fuerza muscular en las cuatro extremidades e hiporreflexia; es dada de alta sin un diagnóstico definitivo. Cuatro meses antes del ingreso nota tumoración parotídea y maxilar derecha, dolorosa, que progresa hasta dificultar la lateralización del cuello. Un mes antes de ingresar presenta vómitos alimentarios y episodios de cefalea occipital, además de disminución de la agudeza visual y desviación de la mirada del ojo derecho. Pocas horas antes de ingresar pierde el conocimiento y no responde a estímulos verbales por lo que es traída a emergencia para su internamiento.
Antecedentes:
Gestaciones: 4 Abortos: 2
Examen Físico:
Presión arterial: 100/60 mmHg Frecuencia cardiaca: 130 x min.
Frecuencia respiratoria: 18 x min.
Paciente en regular estado general, adelgazada, hidratada. Piel: palidez de piel y mucosas. Tejido celular subcutáneo: no hay edemas Ojos: desviación de la mirada al lado izquierdo. Pupilas: 2.5 mm derecha, 5 mm izquierda, poco reactivas. Nariz: secreción purulenta en fosa nasal derecha. Masa sólida de + 8 cm en región parotidea y maxilar derecha. No linfadenomegalias. Tórax y pulmones: murmullo vesicular pasa bien ambos campos pulmonares, no hay rales. Aparato cardiovascular: ruidos cardiacos rítmicos, de buena intensidad, taquicárdicos no soplos. Abdomen: excavado, ruidos hidroaéreos presentes, blando, depresible, no masas, no visceromegalia. Sistema nervioso: Somnolencia, orientada en espacio y persona, hemiparesia izquierda a predominio crural, no signos meníngeos. Anisocoria con desviación de la mirada hacia la izquierda. Babinsky negativo.
Discusión clínica:
Dr. Hugo Umeres
Es una historia clínica con evolución crónica de un año de duración que se inicia con síntomas sensitivos: parestesias y disestesias en el miembro inferior derecho, después se presenta debilidad descendente en esa pierna; y también parestesias en el antebrazo izquierdo y en la pierna izquierda.
Las manifestaciones sensitivas afectan las cuatro extremidades con una distribución mas o menos asimétrica, y además hay disminución de la fuerza muscular e hiporreflexia. ¿Qué se puede concluir de esta información asociada al resultado de la electromiografía?
Que la paciente tiene un compromiso del sistema nervioso con una neuropatía periférica.
En la evaluación clínica de un paciente con neuropatía, debemos realizar un análisis de las diferentes características clínicas para focalizar el diagnóstico (1).
Por ejemplo es necesario conocer la evolución de los síntomas sensitivos, y así podremos diferenciar entre los diferentes tipos de neuropatía:
- Mononeuropatías.
- Mononeuropatías múltiples.
- Polineuropatías simétricas.
Las mononeuropatías se caracterizan por lesión de un solo nervio, ya sea periférico o craneal, ejemplos de una mononeuropatia de pares craneales es la parálisis facial, o la parálisis aislada de una rama del motor ocular común, que se va a manifestar como ptosis palpebral; también hay mononeuropatías de nervios periféricos.
El presente caso presenta múltiples nervios afectados, pero en forma asimétrica, sugiriendo que se trata de lo conocido como mononeuropatía múltiple; las manifestaciones fueron de inicio asimétrico; incluso se puede presentar asimetría en el mismo miembro.
Las polineuropatías simétricas, como por ejemplo la polineuropatia diabética afecta los miembros inferiores de manera ascendente. Las polineuropatias simétricas también podemos categorizarlas en polineuropatias sensitivas, motoras, y mixtas; así como definir si la lesión corresponde a desmielinización o a daño axonal.
Las polineuropatías periféricas simétricas pueden ser desmielinizantes o axonales. ¿Por qué hacemos esta diferenciación? Porque el diagnóstico diferencial de estas es muy amplio y su evaluación muy costosa; por eso utilizamos la clínica en primer lugar y luego la electromiografia para definir entre una neuropatía desmielinizante y una axonal (2).
En la polineuropatía desmielinizante la velocidad de conducción esta por debajo del 30%, en tanto en la axonal la velocidad de conducción suele ser normal y más bien se observan manifestaciones periféricas de denervación tales como ondas positivas o fibrilaciones.
Clínicamente las manifestaciones de la polineuropatia axonal tienden a ser ascendentes como en el ejemplo clásico de la diabetes mellitus, en tanto que en la forma desmielinizante es descendente como suele ocurrir en el síndrome de Guillain – Barre.
En este caso los hallazgos electromiográficos no nos permiten diferenciar entre las diferentes formas de polineuropatía; el médico que realiza el examen usualmente está en capacidad de hacer esa distinción. Pero clínicamente si podemos decir que estamos frente a una mono-neuropatía múltiple.
El diagnóstico diferencial de las mononeuropatias multiples va dirigido en primer lugar a las vasculitis (poliarteritis nodosa y otras) luego en el Perú algunas infecciones, y por último neoplasias y diabetes mellitus.
En el diagnóstico de las polineuropatías desmielinizantes debemos pensar en las causas autoinmunes; en tanto que en las axonales la investigación debe ir dirigida a factores tóxicos, metabólicos (como la deficiencia de vitamina B12, hipotiroidismo, diabetes mellitus, etc). La evaluación de una polineuropatía en su sentido más amplio es muy costosa, en cambio si tenemos la orientación clínica de que sea mononeuropatía múltiple o polineuropatía axonal o desmielinizante el estudio es más directo y menos costoso. A pesar de eso en el 50% de las polineuropatías no podemos establecer un diagnóstico etiológico.
En nuestra paciente la primera electromiografía reveló compromiso mixto, con gran predominio sensitivo; en tanto que la segunda fue descrita con un compromiso sensitivo puro. En presencia de una polineuropatía axonal crónica, sensitiva pura, la posibilidad de que sea una manifestación paraneoplásica se vuelve muy importante (3).
Nos quedamos entonces con la posibilidad de un proceso paraneoplásico cuya lesión primaria se hizo presente posteriormente.
Aún no tenemos el diagnóstico definitivo, pero puede ser una manifestación paraneoplásica, que aparece antes del tumor (en algunos casos dos a tres años antes) (3).
En nuestra paciente la tumoración de la región parotídea apareció cinco meses después de las manifestaciones neurológicas.
Con esta información podemos pensar en una etiología neoplásica ó en un proceso granulomatoso de la parótida; por lo que es importante identificar las neoplásias que afectan a esta glándula.
En presencia de un síndrome paraneoplásico con una neuropatía sensitiva pura en un paciente varón, debemos buscar en primer término un cáncer pulmonar a células pequeñas, y en el caso de ser mujer un cáncer de mama; estas son las más frecuentes. Otras neoplasias que dan estas manifestaciones son las hematológicas, algunos linfomas. Personalmente he visto un caso de un paciente con más de dos años de historia con una polineuropatía sensitiva muy dolorosa, con cinco estudios de electromiografía normales, que en última instancia desarrolló un linfoma primario cerebral. En ese caso se plantea una neuropatía a fibras pequeñas; fibras que conducen el dolor pero que no tienen una traducción electromiográfica (4).
¿Y que sucede después, cuando ya la paciente es internada y claramente hay un cuadro con compromiso del sistema nervioso central? Hay náuseas, vómitos y finalmente alteración de la conciencia, déficit motor caracterizado por una hemiparesia izquierda, anisocoria con desviación de la mirada, que se ha instalado dentro de una polineuropatía. Claramente hay un proceso expansivo intracraneal con hipertensión endocraneana.
Entre los exámenes auxiliares, al inicio tiene un estudio de líquido cefalo- raquídeo, probablemente buscando la etiología de la polineuropatía; en casos de mononeuropatía múltiple no hacemos este estudio pues no da información útil; más bien tomamos una biopsia de nervio porque allí esta el diagnóstico. En las polineuropatias desmielinizantes si esta indicado realizar el estudio de líquido cefalo raquídeo. La impresión diagnóstica nos lleva a tomar la decisión de un examen mas preciso para hacer un diagnóstico.
En su segundo ingreso, el día de la punción lumbar la paciente tiene anisocoria, déficit motor, síntomas de hipertensión endocraneana probablemente en relación con un proceso expansivo intra-craneal, que constituyen una contraindicación clara para realizar este estudio.
En resumen tenemos una polineuropatía, luego aparece una masa en la parótida y esto va seguido por un problema de hipertensión endocraneana por un proceso expansivo. El que la polineuropatía sea sensitiva pura nos lleva a pensar en un síndrome paraneoplásico.
En la radiografía de tórax llama la atención la presencia de calcificaciones; esta paciente viene de Ayacucho de manera que hay que considerar la posibilidad de secuelas de infecciones pulmonares por hongos o tuberculosis; sin embargo, no hay compromiso pleural lo que aleja la posibilidad de que todo el cuadro pueda explicarse por tuberculosis.
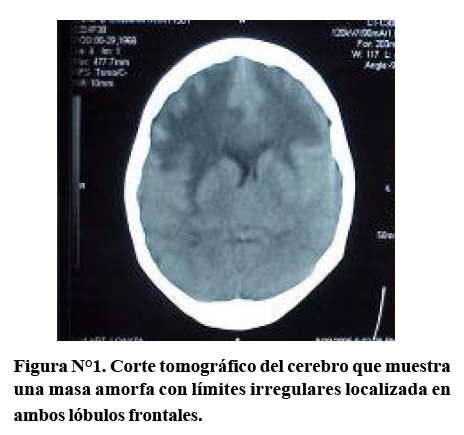
En la tomografía axial computada sin contraste observamos en la parte frontal una masa muy amorfa de límites irregulares con una hipodensidad en la periferia, así como dentro de la propia masa (Figura N°1). Esta lesión está localizada en la zona frontal superior y basal bilateral. En la parte inferior no vemos el cuarto ventrículo; lo cual confirma nuestra afirmación previa de que no debió realizarse una punción lumbar en esta paciente. La lesión está produciendo compresión del ventrículo lateral y agrandamiento con cierto grado de hidrocefalia obstructiva.
En la tomografía con contraste se observa el cuarto ventrículo pequeño, y en la zona anterior se puede ver mejor la masa que es lobulada, con zonas hipodensas, no tiene cápsula y hay clara vascularización. Con estos hallazgos podemos concluir que se trata de una neoplasia, ya sea primaria o metastásica. Las metastásis generalmente ocurren en el territorio de la arteria cerebral media, suelen ser subcorticales; es muy rara la presencia de metástasis en las zonas basales del cerebro.
El origen más común de las metástasis cerebrales es cáncer pulmonar, seguido en frecuencia por lesiones malignas de origen mamario, gastrointestinal y también genito –urinario. No hay evidencia de ninguna de estas neoplasias en esta paciente.
Si planteamos que el tumor primario es de la parotida, debemos considerar que la frecuencia con que estas neoplasias dan metástasis al sistema nervioso central es menor del 1% (5).
Pasando a considerar las neoplasias primarias cerebrales, los meníngiomas son muy vascularizados y los bordes de la lesión son precisos, pues el tumor generalmente esta bien encapsulado, aunque si pueden ir con edema cerebral en la periferie. La historia en estos casos es crónica y muchas veces sin signos de compromiso de pares craneales, pero pueden presentar cefalea y convulsiones. El glioblastoma multiforme es un glioma de grado avanzado que pasa la línea media, y tiene forma de alas de mariposa, presenta zonas de necrosis, son generalmente muy agresivos.
Los síntomas neurológicos de esta paciente no son muy recientes, meses antes tuvo naúseas y vómitos, ahora la masa es grande produciendo complicaciones por efecto del desplazamiento que genera la masa tumoral.
Los linfomas, que no son primarios del sistema nervioso, van a dar compromiso del espacio subaracnoideo y con frecuencia son múltiples. En cambio el linfoma primario que se observa en los pacientes inmuno-suprimidos por SIDA si puede producir lesiones únicas.
Entre las causas inflamatorias podría tratarse de una presentación seudo-tumoral de granulomatosis tuberculosa; la posibilidad de vasculitis la considero alejada pues se manifiesta con lesiones de tipo isquémico.
¿Que otras causas inflamatorias más raras se pueden plantear? Las amebiasis de vida libre; la paciente tiene una descarga nasal purulenta, pero no tiene signo meningeos como los que encontramos en estos pacientes.
En conclusión con los datos que tenemos de la electromiografía, el primer diagnostico a considerar es una polineuropatia de tipo paraneoplasico; con un origen primario pulmonar y más raro de otros orígenes. También estamos planteando la posibilidad de un linfoma que ciertamente puede comprometer la parotida, así como el sistema nervioso central y producir polineuropatia.
Las granulomatosis, particularmente la sarcoidosis que compromete la parotida pueden dar estas imágenes en sistema nervioso, pero suelen dar neuropatías craneales, tales como neuralgias del trigémino o parálisis facial atípica.
Las vasculitis granulomatosas pueden dar imágenes semejantes en el sistema nervioso y comprometer la parotida así como dar manifestaciones de lesión en nervios periféricos, no esta claro si esta paciente tiene o no una mononeuropatía múltiple; hubiera sido importante una biopsia del nervio para dilucidar este diagnóstico. El electromiografista puede tener dificultad para diferenciar entre una polineuropatía o de una fase avanzada de mononeuropatia múltiple. Cuando la mononeuropatía múltiple ha comprometido varios troncos nerviosos se comporta como una polineuropatía; en esos casos se debe interpretar la progresión clínica de las molestias en su inicio, y definir si han sido simétricas o asimétricas. La presentación asimétrica es característica de lesiones vasculíticas.
Otras causas como la mucormicosis, son mucho más raras; se observan en pacientes inmuno comprometidos, por ejemplo en diabéticos descompensados; la paciente tiene una secreción nasal purulenta, sin sangrado; pero las imágenes radiológicas de la lesión cerebral no son las características.
Dr. Cipriani: ¿Podríamos ver cortes bajos en la tomografía para ver senos paranasales ?
Dr. Umeres: En este corte el seno etmoidal es anormal y esto sugiere la posibilidad de una lesión inflamatoria, infecciosa o tumoral a este nivel (Figura N°2). Me gustaría conocer el resultado de la evaluación otorrinolaringológíca.

Dr. Ramos: A la paciente le practicaron una endoscopía que revelo una lesión tumoral en la fosa nasal, que fue biopsiada.
Discusión Patológica:
Dr. Cesar Chian.
A la paciente se le tomaron dos biopsias en el intervalo de un año. La primera biopsia es de nervio sural y la segunda de una tumoración nasal. Con las técnicas que utilizamos actualmente nosotros podemos contribuir mediante el estudio de biopsias de nervio sural al diagnóstico de enfermedades como poliarteritis nodosa y otros tipos de vasculitis, neuropatías asociadas con depósitos como amiloidosis o diabetes y lepra, principalmente. Para el diagnóstico de neuropatías desmielinizantes se requiere de procedimientos especiales y de microscopía electrónica que son realizados en centros especializados.
En la biopsia de nervio periférico (Figura N°3) los únicos hallazgos observados fueron presencia de mínima cantidad de linfocitos dispersos entre las fibras nerviosas con discretos focos de degeneración mielínica. Los hallazgos fueron inespecíficos y no diagnósticos.

La segunda biopsia fue tomada durante una rinoscopía en la que se visualizó una proliferación neoplásica en la cavidad nasal. En esta biopsia (Figura Nº 4) se observa una gran proliferación celular alrededor de los vasos sanguíneos con extensa necrosis tumoral en las zonas alejadas de los vasos. Las células neoplásicas están dispuestas en mantos y tienen núcleos hipercromáticos y pleomórficos. Estos hallazgos son característicos de un carcinoma.

Las enfermedades que tienen localización intranasal son muy variadas y constituyen un reto diagnóstico (Tabla N°1).

La principal diferencia entre carcinoma sinonasal y nasofaríngeo, además de su diferente origen anatómico está en que el segundo es mas frecuente en el Asia y muestra una asociación muy fuerte con infección por virus de Epstein-Barr.
El carcinoma sinonasal puede ser de tipo escamoso, transicional, verrucoso, adenocarcinoma, neuroendocrino, entre otros; mientras que los tipos histológicos de carcinoma nasofaríngeo son: carcinoma escamoso queratinizante, carcinoma escamoso no queratinizante o carcinoma indiferenciado con infiltrado linfocítico no neoplásico (6). El mal pronóstico de los carcinomas sinonasales y nasofaríngeos está relacionado con su diagnóstico usualmente tardío, porque la localización anatómica de los tumores no suele relacionarse con sintomatología en fases tempranas de la enfermedad (7).
Se realizó inmunohistoquímica e hibridación in-situ para EBV. El tumor fue positivo para pankeratina (patrón perinuclear punteado), sinaptofisina y enolasa neurona específica y CD56, y la hibridación in-situ para EBV fue negativa. Los resultados de la inmunohistoquímica permitieron clasificar esta neoplasia como carcinoma neuroendocrino pobremente diferenciado.
Las neuropatías paraneoplásicas son un grupo de trastornos del sistema nervioso significativamente relacionados con neoplasias malignas, después de haber excluido enfermedad metastásica, complicaciones iatrogénicas, infecciones oportunistas promovidas por inmunosupresión y desórdenes metabólicos o deficiencias nutricionales relacionadas con la malignidad (8). Los síndromes paraneoplásicos que compromente el sistema neuromuscular se clasifican en miopatías inflamatorias, bloqueos neuromusculares (Lambert Eaton), neuropatías periféricas y mielopatías necrotizantes paraneoplásicas (8). La mayor parte de neuropatías paraneoplásicas están asociadas a cáncer de células pequeñas (neuroendocrino) de pulmón y son neuropatías sensoriales subagudas causadas por una injuria a los cuerpos neuronales sensoriales de los ganglios de las raíces nerviosas posteriores mediada por anticuerpos anti-Hu (9,10). Estos mismos anticuerpos también se han descrito en relación con cánceres del tubo digestivo, próstata, mama, vejiga, riñón, páncreas, testículo y ovario. Se desarrolla neuropatía periférica paraneoplásica en 60 a 95% de pacientes con cáncer y títulos elevados de anticuerpos anti-Hu (11). Otros anticuerpos relacionados con neuropatías paraneoplásicas, son las anticuerpos tipo 2 contra el citoplasma de las células de Purkinje, anticuerpos contra la proteína 5 mediadora de la respuesta de la colapsina y anticuerpos contra los receptores nicotínicos de acetilcolina (12).
Podemos concluir que la paciente presentó una polineuropatía como síndrome paraneoplásico con manifestaciones clínicas que precedieron en un año al desarrollo del tumor endonasal; a pesar de no haber realizado estudios para detectar anticuerpos anti-Hu. Los hallazgos en la biopsia de nervio sural tampoco son diagnósticos en este caso, pero la presencia de ocasionales células inflamatorias y la degeneración mielínica si reflejan el daño primario del nervio periférico.
Diagnóstico final:
Carcinoma neuroendocrino pobremente diferenciado de la región nasal con polineuropatía paraneoplasica.
AGRADECIMIENTO: Al Dr. Carlos Torres Cabala, del Instituto Nacional del Cáncer de los Estados Unidos por su gentil colaboración en la revisión del caso y las pruebas de inmunohistoquímica e hibridización in-situ.
REFERENCIAS BIBLIOGRÁFICAS
1.Bromberg MB. An Approach to the Evaluation of Peripheral Neuropathies. Sem Neurol 2005; 25: 153-159. [ Links ]
2.Asbury AK. Approach to the patient with Peripheral Neuropathy. Harrison's principles of internal medicine. 16th edition. Mc Graw Hill; 2005. p. 2500 – 2510. [ Links ]
3.Darnell RB, Posner JB. Paraneoplastic syndromes involving the nervous system. N Eng J Med 2003; 349: 1543 – 1554. [ Links ]
4.Mendell JR, Sahenk Z. Painful sensory neuropathy. N Eng J Med 2003; 348: 1243 – 1255. [ Links ]
5.Debevec M. Management of patients with brain metastases of unknown origin. Neoplasma 1990; 37: 601– 606. [ Links ]
6.Rosai J. Rosai and Ackerman´s Surgical Pathology. 9th Ed. Philadelphia: Mosby; 2004. p. 305-334. [ Links ]
7.Robins y Cotran. Patología estructural y funcional, 7ma edición; 2005. p. 789. [ Links ]
8.Poirier J, Gray F, Egcourolle R. Manual of Basic Neurophatology, 3th edition. 1990. p.174. [ Links ]
9.Lucchinetti Cf, Kimmel DW, Lennon VA. Paraneoplastic and oncologic profiles of patients seropositive for type 1 antineuronal nuclear autoantibodies. Neurology 1998; 50: 652 – 57. [ Links ]
10.Camdessanche JP, Antoine JC, Honnorat J, et al. Paraneoplastic peripheral neuropathy associated with anti – Hu antibodies: a clinical and electrophysiological study of 20 patients. Brain 2002; 125: 166 – 75. [ Links ]
11.Dalmau J, Graus F, Rosenblum MK, Posner JB. Anti-Hu-associated paraneoplastic encephalomyelitis/sensory neuropathy: a clinical study of 71 patients. Medicine 1992; 71:59-72. [ Links ]
12.Freeman R. Autonomic peripheral neuropathy. Lancet 2005; 365:1259-70 [ Links ]

