










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Medica Herediana
versión impresa ISSN 1018-130Xversión On-line ISSN 1729-214X
Rev Med Hered v.20 n.2 Lima abr./jun. 2009
Conversatorio clínico patológico en el Hospital Nacional Arzobispo Loayza_2009-02.
Clinical Case at the Hospital Nacional Arzobispo Loayza- 2009-2.
Editor Responsable: Dr. Enrique Cipriani Thorne1
Editores Asociados: Dra. Gloria Bravo Muro2 ,Dr. Jorge Casas Castañeda1,Dr. Abdías Hurtado Arestegui1, Dra. Giovanna Rodríguez Lay3
Discusión Clínica: Dra. Ana Cecilia Olascoaga4
Discusión Patológica: Dra. Mercy Jhong5
Médico Residente: Dr, Jaime Antonio Retamozo Arias6
Colaboración : Dr. Braulio Valencia Arroyo7
1
Profesor Principal de Medicina. Facultad de Medicina Alberto Hurtado Universidad Peruana Cayetano Heredia. Lima, Perú2
Profesor Auxiliar de Patología. Facultad de Medicina Alberto Hurtado Universidad Peruana Cayetano Heredia. Lima, Perú3
Profesor Asociado de Medicina. Facultad de Medicina Alberto Hurtado Universidad Peruana Cayetano Heredia. Lima, Perú4
Médico Asistente de Medicina. Hospital Nacional Arzobispo Loayza. Lima, Perú5
Médico Asistente de Hematología. Hospital Nacional Arzobispo Loayza. Lima, Perú6
Residente de Medicina de Postgrado de la Universidad Nacional Mayor de San Marcos. Lima, Perú7
Interno de Medicina. Facultad de Medicina Alberto Hurtado de la Universidad Peruana Cayetano Heredia. Lima, Perú
Enfermedad actual
Paciente mujer de 18 años, natural de Huancavelica y procedente de Ayato, Huancavelica; soltera.
Cuatro meses antes de su ingreso presenta dolor en codos y hombro derecho que limita el sueño. Además presenta disminución de la fuerza muscular en miembros inferiores a predominio proximal que aumenta progresivamente, posteriormente compromete los miembros superiores. También hay caída del cabello, hiporexia, sequedad bucal y pérdida progresiva de peso de 42 a 36 Kg. Refiere rigidez articular matutina de miembros superiores menor de 30 minutos de duración, frialdad distal y coloración azulada de uñas y dedos en periodos de frio.
Un mes antes del ingreso inicia sensación de alza térmica acompañada de escalofríos, sin predominio de horario, el dolor articular se intensifica a predominio de codos, hombros y articulaciones coxofemorales.
La semana previa al ingreso limita su deambulación de manera importante, pudiendo desplazarse solo con ayuda. Cuatro días antes del ingreso viene a Lima y acude a una clínica particular donde le refieren que presenta anemia y le transfunden un paquete globular.
Antecedentes:
No hepatitis, TBC, tifoidea, asma, brucelosis, hipertensión arterial y Diabetes mellitus. No cirugía y ni hospitalizaciones previas. Gestaciones: ninguna. Usa paracetamol esporádicamente y un complejo vitamínico hace 2 meses por 3 semanas. Una hermana falleció de "Cáncer a los huesos". Niega contactos TBC.
Examen Físico:
PA: 130/80 mm Hg, Frecuencia cardiaca: 108 x min, Frecuencia respiratoria: 26 x min, Temperatura: 40º C.
Regular estado general y de hidratación, mal estado de nutrición. Despierta, en decúbito dorsal obligado, conectada con el entorno. Piel: Caliente, pálida, humedad disminuida, no rash, no ictericia. Tejido celular subcutáneo: disminuido, no edemas. Sistema linfático: múltiples micro adenopatías cervicales, inguinales y poplíteas. Sistema osteomioarticular: Dolor a la digito presión en D4, columna lumbosacra, en hombro derecho y en codos. Cabeza: normocéfala. Escleras azules. Lengua papilada; labios secos. Cuello: no se palpa tiroides, múltiples adenopatías de aproximadamente 0,5 cm. Mamas: normales. Tórax y pulmones: amplexacion conservada, murmullo vesicular pasa bien en ambos campos pulmonares, no ruidos agregados. Sistema cardiovascular: ruidos cardiacos rítmicos, de buena intensidad, presenta soplo sistólico II/VI en toda la región precordial. Abdomen: ruidos hidroaéreos presentes, blando y depresible; hígado palpable a nivel de reborde costal; dolor en hipocondrio derecho. Genitourinario: puño percusión lumbar y puntos renoureterales no dolorosos. Genitales: normales. Neurológico: lucida y orientada en las tres esferas; disminución de la fuerza muscular proximal; sin alteraciones en la sensibilidad; no signos meníngeos; signo de Babinsky negativo; reflejos osteotendinosos patelares incrementados.
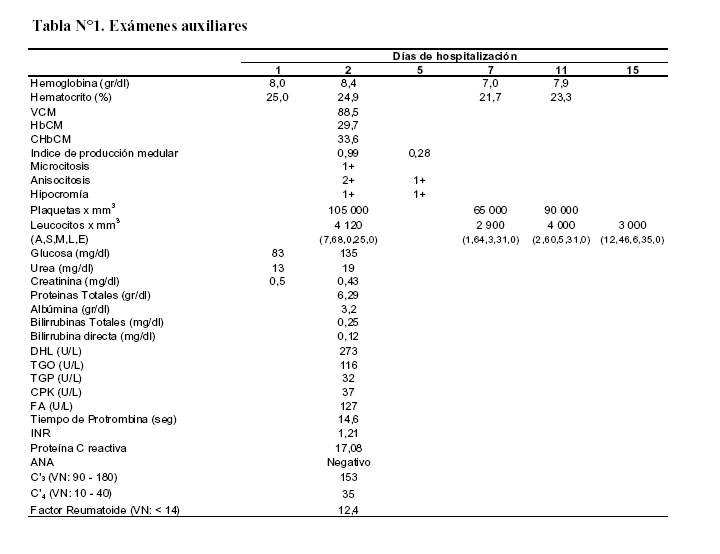
Exámenes auxiliares:
Los exámenes de laboratorio sanguíneos y su evolución se muestran en la tabla N°1.
La ecografía abdominal fue normal. La proteinuria en 24 horas fue 387 mg y el examen de orina mostró: pH 6,5 densidad 1005, leucocitos 2 – 5 x campo, hematíes 0 – 2 x campo, células epiteliales numerosas y gérmenes 1+.
La electromiografía demostró velocidad de conducción nerviosa normal, negativa para polineuropatía.
El estudio completo para brucella y el mielocultivo, fueron negativos.
Se realizó el procedimiento de diagnóstico.
Discusión clínica:
Dra. Ana Cecilia Olascoaga
Se trata de una mujer joven de 18 años, soltera, de ocupación niñera y procedente de Ayato; una ciudad de la sierra en la que casi todos los pobladores son indígenas y la mayoría de personas se dedica a la agricultura. Esto es importante por dos motivos: En primer lugar si la paciente procediera de Lima o de una ciudad más grande podría tener mayor riesgo de padecer de infecciones como VIH y en segundo lugar si la paciente se dedicaría a la agricultura, el contacto probable con pesticidas la pone en riesgo de enfermedades asociadas a su uso (1,2).
En cuanto a los otros antecedentes no hay información sobre relaciones sexuales y niega el contacto con personas con TBC. El único dato saltante es que la hermana falleció por un cáncer del cual no tenemos descripción, siendo este dato muy importante, conocida la influencia de la herencia en algunas neoplasias como ciertas leucemias (3).
La enfermedad actual es de curso crónico y los signos y síntomas principales son los siguientes: dolor y rigidez articular, debilidad muscular, pérdida de peso, linfoadenopatías, fiebre, alopecia, escleras azules y sequedad bucal. A esto se suman los hallazgos de laboratorio, entre ellos: proteinuria, enzimas hepáticas alteradas y el hallazgo hematológico final. Vamos a hacer un análisis de los síntomas principales y así tratar de llegar a un diagnóstico.
Al hablar de dolor debe evaluarse la localización, duración, intensidad, signos asociados, ritmo y evolución. En la paciente el dolor se localiza en codos, hombro derecho y articulaciones coxofemorales y su curso altera el sueño. Al examen físico se encuentra también dolor en D4 y en la columna lumbosacra y se evidencia dolor a la digito presión en hombro derecho y codos.
El dolor podría tener un origen articular, muscular, neuropático u óseo. Si la causa del dolor es la articulación y el tejido peri articular, éste se exacerba con los movimientos; es raro que haya dolor articular sin movimiento y hay algunos signos que permiten definirlo mejor. El dolor muscular suele ser profundo, mal localizado y empeora con el movimiento y el ejercicio. El dolor neuropático es continuo, mal localizado, urente y tiene reagudizaciones paroxísticas que no dependen del movimiento. El óseo es también un dolor constante pero las características dependen de la causa que lo produce. En la historia clínica solo se precisa la localización, y esto no permite definir con exactitud la fuente del mismo. Es decir, podría indicarse un dolor en la rodilla y el problema estar realmente en la cadera.
La duración y el inicio también son importantes: de inicio súbito en el lumbago y gradual en artritis reumatoide, artrosis y enfermedades óseas. La paciente tiene un dolor gradual y bien localizado.
La descripción de la intensidad del dolor tiene una connotación especial según la personalidad de cada paciente, pero el dato que si indica un dolor intenso es la interferencia con el sueño y el trabajo, en este caso debería considerarse que la paciente presenta un dolor muy intenso.
Los signos que se evalúan en un compromiso articular son: tumefacción, rubor, calor, derrame articular, edema, deformidad, actitud viciosa en la posición y alteración de la movilidad; ninguno de estos signos está presente.
El dolor mecánico aumenta con la actividad y calma con el reposo, mientras que el inflamatorio persiste en la noche, el dolor psicógeno no sigue ningún patrón, como en la fibromialgia que varía según las emociones, y el dolor neoplásico, que es constante, cede poco con los analgésicos al igual que el dolor neuropático.
En este caso el dolor podría ser inflamatorio o neoplásico. La evolución nos podría ayudar en caso de ser aditivo, migratorio o autolimitado.
Es importante el dato de la existencia de dolor a la digito-presión ya que permite definir si el dolor se origina a nivel articular, óseo o de masas musculares o tendíneas. Las lesiones articulares las alejamos ya que no hay hallazgos de signos articulares. La presencia de masas musculares también se aleja. Con todo esto se concluye que se trata de un dolor óseo.
Las causas de dolor óseo son diversas: metabólicas, tumorales e infecciosas. Osteomalacia se aleja porque la paciente no presenta deformidades óseas, desnutrición ni diarrea crónica. La enfermedad de Paget es localizada y causa deformidad. La osteoporosis produce dolor en columna vertebral, el hiperparatiroidismo produce cálculos renales, trastornos de conciencia y ocasionalmente crisis de pseudogota, nada de lo cual presenta la paciente.
Las enfermedades tumorales primarias y metastásicas pueden ser consideradas, al igual que infecciones como TBC y brucelosis. Las displasias óseas como osteopetrosis, osteomieloesclerosis, displasia diafisiaria progresiva y displasia fibrosa son alteraciones que se presentan desde la infancia y producen deformidades y tendencia a las fracturas nada de lo cual esta presente.
La evaluación de debilidad muscular se hace simultáneamente con el tono muscular, la presencia de atrofia, los reflejos osteotendinosos, la presencia de fasciculaciones, la distribución de la debilidad y la presencia o ausencia de signo de Babinsky ( Tabla Nº2)(4).
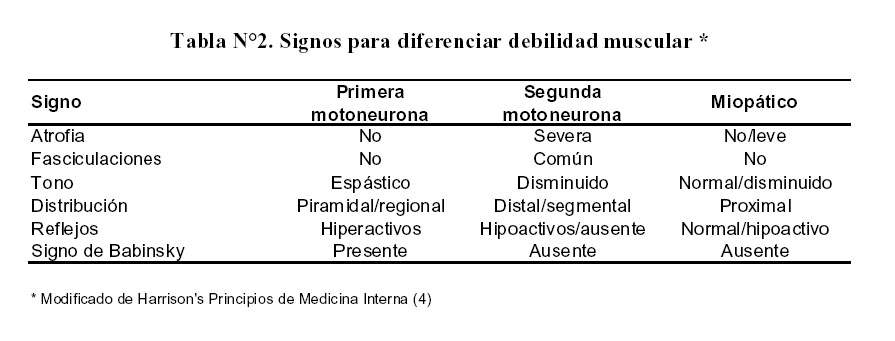
Signos para diferenciar debilidad muscular
La debilidad muscular en la paciente se manifiesta como disminución de la fuerza en miembros inferiores a predominio proximal, que aumenta y progresa posteriormente a miembros superiores, hasta que finalmente solo puede caminar con ayuda. En el examen físico se evidencia disminución de la fuerza muscular proximal.
El tono muscular permite definir si el compromiso es de primera neurona, caracterizada por espasticidad o rigidez, como en los síndromes extra piramidales y el Parkinson; o paratonía en las enfermedades del lóbulo frontal. Cuando el tono esta disminuido o normal como en el caso de esta paciente el desorden se encuentra en la segunda motoneurona o en las fibras musculares que inerva.
En nuestro caso, la distribución de la debilidad es proximal, el signo de Babinsky esta ausente, no hay fasciculaciones ni atrofia y los reflejos osteotendinosos están aumentados solo en la pierna derecha, por ello estamos ante un patrón miopático, aunque con reflejos osteotendinosos incrementados de manera asimétrica lo cual es un patrón inusual y podría ser un error en la exploración; o tener otro significado, como un problema vertebral sobre agregado, que afecte la segunda motoneurona; sin embargo existe un estudio de velocidad de conducción y electromiografía que descartan un compromiso periférico, al igual que de primera motoneurona y miopático.
Como causas de miopatía tenemos a las enfermedades de fibras musculares y de la unión neuromuscular como la miastenia gravis; esta última se puede descartar por las características que tiene la paciente. La dermatomiositis y polimiositis tienden a presentar debilidad, CPK elevada y electromiografía con patrones típicos por lo cual éstas se alejan. Las miopatías hereditarias se presentan tempranamente y no explican el cuadro clínico de nuestra paciente. Las enfermedades endocrinológicas como hipo o hipertiroidismo podrían explicar el cuadro de debilidad pero no el dolor. Las enfermedades sistémicas por pérdida muscular severa podrían ser una causa en el caso de la paciente, la ingesta de tóxicos no está presente en la historia y no existe un registro de probable hipercalcemia. Creo que la paciente tiene una pérdida muscular severa debida al cuadro de fondo.
La pérdida de peso ha sido de 14%, se asocia a hiporexia y no hay edemas. Las causas de perdida de peso se clasifican en tres grupos: aumento del consumo de energía, aumento de la pérdida de energía por heces u orina y disminución de la ingesta calórica. Cuando hay incremento del gasto el paciente come lo usual o más de lo usual lo cual no sucede en esta paciente. Cuando se debe a la pérdida de energía tanto por orina como por heces el paciente come normalmente o más de lo usual y se ve en pacientes con diabetes o en enfermedades que producen diarrea; estas situaciones no suceden en nuestra paciente, por lo cual nos quedamos con la disminución de la ingesta calórica que parece ser por pérdida del apetito.
Las causas de anorexia son diversas: neoplasias, infecciones (HIV, TBC, endocarditis), hipercalcemia, uremia, enfermedades gastrointestinales obstructivas, insuficiencia adrenal, enfermedades crónicas (ICC, EPOC, cirrosis), enfermedades autoinmunes, depresión y Alzheimer. Con todo ello nuestras principales sospechas son las causas neoplásicas y las infecciosas; las enfermedades autoinmunes y las endocrinológicas no parecen corresponder a nuestro caso.
La paciente tiene múltiples adenopatías axilares, inguinales y poplíteas. Cuando se evalúan pacientes con linfoadenopatías tenemos que ver su extensión, tamaño, textura, movilidad y si presentan esplenomegalia asociada. El compromiso localizado se ve en casos de neoplasias e infecciones locales. Se consideran generalizadas si hay compromiso de 3 ó más áreas linfáticas como en el caso de nuestra paciente. Las enfermedades asociadas con linfoadenopatía generalizada son: mononucleosis infecciosa, toxoplasmosis, HIV, lupus eritematoso sistémico, enfermedad mixta del tejido conectivo, leucemias y linfomas.
Los ganglios pequeños (<1cm) son por lo general benignos y cuanto mayor es su volumen mayor es la probabilidad de que sean malignos. La textura se refiere a si son móviles y blandos, ó fijos y duros como en el caso de las neoplasias. La presencia de dolor indica inflamación asociada. La esplenomegalia sugiere cuadros sistémicos: mononucleosis infecciosa, linfoma, leucemia, LES, sarcoidosis y toxoplasmosis. La conclusión es que se trata de una linfoadenopatía generalizada, probablemente neoplasia hematológica o alguna infección.
La alopecia puede ser cicatricial o no cicatricial; la diferencia es que la última es reversible. La cicatricial se asocia a inflamación, fibrosis con pérdida de folículos pilosos y es causada por entidades como liquen plano, lupus cutáneo, escleroderma lineal, sarcoidosis y metástasis cutáneas. La no cicatricial es causada por enfermedades cutáneas primarias (telógeno efluvium: por estrés o enfermedad importante, alopecía androgénica, alopecía areata y tiña capitis), exposición a drogas (que inducen a telógeno efluvium), LES y sífilis y el adelgazamiento difuso del cabello: hipertiroidismo, hipopituitarismo, infección por HIV, deficiencias proteicas o por hipotiroidismo. Las causas cicatriciales se descartan por completo; de igual manera las no cicatriciales debidas a alteraciones metabólicas y carenciales, así como el LES por el ANA negativo; solamente queda el telógeno efluvium debido al deterioro general causado por la enfermedad de fondo.
La xerostomía puede ser fugaz o duradera. Las causas persistentes son variadas: diabetes mellitus, diabetes insípida, uremia, hipotiroidismo, mucoviscidosis, síndrome de Sjögren y síndrome de Mikulicz. En nuestra paciente las posibilidades se limitan a las dos últimas.
El síndrome de Mikulicz se caracteriza por una infiltración de las glándulas salivales por ejemplo en las leucemias. El síndrome de Sjögren en su forma secundaria se asocia con otras manifestaciones que no están presentes. La conclusión es que se trata de un síndrome de Mikulicz o de síndrome de Sjögren primario.
La rigidez matutina indica inflamación articular (artritis reumatoide vs otras artritis) o deformidad (artrosis). No existen datos en el examen físico que orienten a definir este problema, así como tampoco hay criterios de síndrome reumatoide por lo cual no se incluye en el diagnóstico diferencial.
La frialdad distal con cambio de coloración la asociamos con el fenómeno de Raynaud y la acrocianosis. El primero es causado por un reflejo simpático exagerado o mayor sensibilidad al mismo, es episódico y asociado al frío y tiene tres fases características: palidez, cianosis y rubor. La acrocianosis por otro lado la presentan mujeres jóvenes, empeora con el frío y no tiene las tres fases mencionadas.
La historia refiere que la paciente tiene escleras azules, que es algo que puede encontrarse en niños y también en mujeres. Cuando la esclera es muy delgada puede tomar el color de la úvea, esto se da en la osteogénesis imperfecta; existen otras causas: anemias sideropénicas, anquilostomiasis, procesos reumáticos tratados con corticosteroides, síndrome de Ehlers-Danlos, seudoxantoma elástico y miastenia gravis; por último podría ser un hallazgo sin mayor implicancia.
La fiebre en la paciente tiene un mes de duración puede tener un origen infeccioso, autoinmune o neoplásico y podría ser también una infección sobre agregada en alguien crónicamente enfermo.
En el estudio de una paciente con proteinuria se debe considerar que ésta puede verse en casos con fiebre o en enfermedad aguda debido a cambios glomerulares hemodinámicos. Si la proteinuria es persistente puede ocurrir por sobreproducción de proteínas de bajo peso molecular: la proteinuria de Bence-Jones, mioglobinuria, hemoglobinuria, lisozimuria (leucemias mielomonocíticas), â2-microglobulinuria (neoplasias); pérdida a nivel glomerular en enfermedades glomerulares; y por último, la proteinuria puede ser tubular en caso de enfermedades hereditarias (Enfermedad de Wilson), lesión tóxica (cadmio, plomo), metabólicas (hipokalemia), síndrome de Fanconi, nefritis crónica intersticial. La causa más probable en nuestra paciente es por sobreproducción.
Nuestra enferma tiene la TGO elevada y la TGP normal. La TGO es un marcador de compromiso de hígado, corazón y músculo y la TGP principalmente de lesión hepática. Generalmente las dos van juntas, solo hay algunas condiciones en las cuales no ocurre esto: hepatitis alcohólica y la enfermedad de hígado graso asociado al embarazo. Si la TGO está elevada aisladamente sugiere un problema no necesariamente hepático. La FA está también elevada y esto ocurre en el compromiso hepático, óseo, intestinal y placentario.
Finalmente y en la base de todo se encuentra el compromiso hematológico: la hemoglobina al ingreso está disminuida y tiene constantes corpusculares normales; el mismo día tiene dos frotises de sangre periférica, uno con microcitosis, anisocitosis e hipocromía y el otro con los reticulocitos disminuidos, la serie blanca disminuida y plaquetas disminuidas. En la evolución los leucocitos están disminuidos y siguen cayendo al igual que las plaquetas.
En conclusión tenemos una anemia normocitica, normocrómica y pancitopenia; los reticulocitos están en 2,8% y corrigiendo el IPM su valor es bajo, se trata de una pancitopenia por baja producción medular. Con dichos hallazgos se debe hacer un estudio de médula ósea.
De tratarse de una anemia aplásica, esta solo da síntomas debido a las citopenias y no un cuadro tan florido como en la paciente; los trastornos mieloptísicos son causa importante; el mieloma múltiple se aleja por la edad, las anemias mielodisplásicas y diseritropoyéticas se presentan por lo general en niños; nos quedamos con anemia por mieloptisis. El cuadro clínico en la mieloptisis varía según la etiología, pueden ser síntomas asociados a la enfermedad subyacente, o dolor e inflamación de los huesos afectados, fracturas patológicas, hipercalcemia, náuseas, síntomas de anemia, debilidad muscular y en el frotis de sangre periférica encontramos anisocitosis, poiquilocitosis, dacriocitosis y glóbulos nucleados. En general las leucemias crónicas no producen tanta pancitopenia comparadas con las leucemias agudas.
Las leucemias comienzan en la médula y luego van a los ganglios al contrario de los linfomas; estos últimos muestran más compromiso orgánico y luego hematológico.
Otras causas a tener en cuenta son: la fibrosis de médula ósea, la policitemia vera, la leucemia de células peludas que van con una gran esplenomegalia; además tumores malignos metastásicos como de pulmón, mama, próstata, etc. Dentro de las infecciosas debemos tener en cuenta: la TBC, la brucelosis e infecciones fúngicas. Se descarta la brucelosis ya que la paciente cuenta con estudios serológicos y cultivos negativos para brucella. La sarcoidosis es una enfermedad poco prevalente en el Perú y no encuentro asidero para considerar las enfermedades de depósito.
En conclusión, mis probabilidades diagnósticas en orden de importancia son: Leucemia aguda (linfoblastica vs mieloide), TBC multisistémica con compromiso óseo diseminado y en tercer término carcinoma metastásico de origen desconocido.
Discusión patológica
Dra. Mercy Jhong
El servicio de Hematología evaluó a la paciente encontrando tres problemas principales: dolor articular, fiebre y bicitopenia. Entre las posibilidades diagnósticas consideramos a la enfermedad del tejido conectivo y una neoplasia hematológica; realizándose por tal razón la serología para anticuerpos antinucleares que resultó negativo y se planteó el estudio de médula ósea.
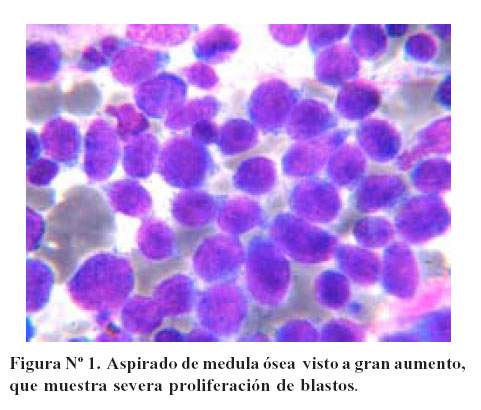
Se realizó el aspirado de médula ósea del esternón, consiguiendo una buena muestra. El estudio del frotis de médula ósea reveló a menor aumento una celularidad incrementada, con aspecto homogéneo y muy escasa cantidad de tejido graso. A mayor acercamiento se observó la infiltración medular por células blásticas representando más del 70% de la celularidad total. Con estos hallazgos se diagnostica Leucemia Aguda ( Figura Nº1).
La progresión de la hematopoyesis normal nos sirve para entender el origen de la leucemia aguda: la célula madre, pluripotente, tiene capacidad de autorenovación y de autodiferenciación en precursores mieloides, los que a su vez dan lugar a células más diferenciadas que finalmente darán origen a las células sanguíneas que observamos en un frotis de sangre periférica. Cuando las células pluripotentes ocupan la médula ósea y tienen capacidad de dividirse pero no de diferenciarse, se presenta como consecuencia una leucemia aguda.
Normalmente existen proto-oncogenes que mantienen el equilibrio entre la persistencia celular y la apoptosis. Al ocurrir mutaciones en puntos específicos de dichos proto-oncogenes, ocurre la formación de oncogenes que determinan la proliferación celular desordenada. Por otro lado, existen genes supresores de tumores que tienen como función detener estos procesos, pudiendo ser también afectos de mutaciones.
Las leucemias y neoplasias en general tienen un origen clonal, esto significa que al menos son necesarias dos mutaciones independientes para que sea posible desarrollar la enfermedad. Por ejemplo, si en una célula progenitora ocurre en algún momento de su división una mutación, esta se presentará en la célula originada y si luego ocurre una segunda mutación en esta segunda célula; cuando prolifere por segunda vez dará lugar a una célula neoplásica.
La etiología de las leucemias no ha sido bien definida hasta el momento y se han planteado varios factores asociados en su desarrollo como bencenos, pinturas, pesticidas, etc; así como, algunas enfermedades genéticas como el síndrome de Down, quienes tienen una mayor probabilidad de desarrollarla. Sin embargo, en la actualidad no es posible determinar un agente causal definitivo.
La leucemia aguda del adulto en el 80% (5) es mieloide. Las leucemias mieloides agudas (LMA) aumentan su incidencia con la edad y las linfociticas presentan 2 picos etarios: menores de 5 años y otro en los adultos mayores. La presentación clínica está determinada por 2 factores: la disminución de la hematopoyesis normal debida a la mieloptisis resultando en una insuficiencia medular, lo que da lugar a citopenias que se manifiestan como palidez, cansancio y fiebre.
La otra forma de presentación es por la infiltración neoplásica a otros órganos dando síntomas como dolor articular, dolor óseo; este último es frecuente en la leucemia linfocitica aguda y muchas veces puede ser el debut de la enfermedad al igual que las linfoadenopatias y la esplenomegalia.
Además de estas manifestaciones generales se pueden encontrar: el compromiso multiorgánico; en la piel pueden encontrarse placas violáceas asintomáticas conocidas como leucemia cutis, debido a la presencia de mieloblastos en la dermis profunda; el síndrome de Sweet, un cuadro paraneoplásico dado por la infiltración de neutrófílos maduros en la dermis superior causando gran dolor; el cloroma es otra manifestación constituida por el engrosamiento violáceo de la piel debida a una acumulación de células, al presionar se torna verde debido a la presencia de las células mieloides, puede ocurrir en otros órganos y su presencia confiere un peor pronóstico.
El compromiso gastrointestinal usual es la hiperplasia gingival con sangrado y dolor de las encías debido a infiltración, esto ocurre con mayor frecuencia en determinados tipos de leucemia aguda como M3, M4. El compromiso gingival también puede ocurrir luego de la quimioterapia. La leucemia aguda puede debutar con gangrena de Fournier y es importante ya que el síntoma principal muchas veces es solo el dolor durante la defecación. Otra forma poco usual de manifestación a nivel del tubo digestivo es la enterocolitis necrotizante que se presenta como obstrucción intestinal. También puede presentarse la neumatosis como complicación de la quimioterapia.
El corazón también puede afectarse por infiltración neoplásica en cualquier localización y el compromiso más común ocurre durante el uso de quimioterápicos como la antraciclina y la neomicina. La toxicidad cardiovascular no suele ser inmediata y es dosis dependiente (6).
El compromiso ocular es también posible y puede haber sangrado en la retina. El pulmón puede comprometerse en casos en los que hay gran número de blastos circulando lo cual puede determinar la formación de émbolos tumorales que causan leucostasis. La leucostasis también puede comprometer el cerebro causando un cuadro similar al de una trombosis. El compromiso meníngeo también es frecuente por infiltración tumoral, especialmente en la leucemia de etiología linfoide (7).
Las alteraciones del medio interno son variadas pero lo mas frecuente es el síndrome de lisis tumoral que se debe a la liberación de cationes intracelulares en el espacio intravascular debido al alto recambio celular o posterior a la quimioterapia (8).
El diagnóstico se hace de manera presuntiva con el examen físico y un frotis de sangre periferica en el que se observan blastos, aunque no siempre están presentes. El hemograma puede ser muy variable, puede existir leucopenia como manifestación de mieloptisis, leucocitosis; esta última generalmente no tan intensa como en la leucemia mieloide crónica y puede ser la manifestación de la presencia de blastos en sangre periférica. El 10% de las leucemias pueden tener un hemograma normal. En el contexto de la paciente con leucocitos normales, con trombocitopenia y sin blastos en sangre periférica está indicado el estudio de médula ósea para llegar al diagnóstico. Se necesita ver más del 20% de blastos en la médula ósea para el diagnóstico de leucemia aguda.
Existen coloraciones para diferenciar si la leucemia es de estirpe mieloide o linfoide así como anticuerpos monoclonales para detectar el inmunogenotipo. Con estas herramientas diagnosticas, el grupo Franco-Americano-Británico ha establecido 8 tipos de Leucemia Mieloide Aguda (LMA) y 3 tipos de LLA (Leucemia Linfocitica Aguda). La citogenética está indicada para establecer además el pronóstico; sin embargo existe un porcentaje de casos en que la leucemia no puede ser clasificada.
En cuanto al tratamiento, éste se debe realizar lo más pronto posible; se consideran condiciones de extrema urgencia como un conteo de blastos muy altos en sangre periférica o la presentación de coagulación intravascular diseminada (CID), situación frecuente en determinados tipos de leucemia aguda como M3.
Los objetivos del tratamiento son: Obtener remisión completa y mantener dicha remisión. Existen criterios para definir remisión; en nuestro caso no fueron establecidos ya que la paciente fue transferida al Instituto Nacional de Enfermedades Neoplásicas (INEN).
Existen predictores de respuesta: 1) los pacientes mayores tienen peor pronóstico al igual que los que tienen gran carga tumoral periférica; 2) los que la presentan posterior a un síndrome mieloproliferativo, o mielodisplasia (leucemia aguda secundaria) y 3) marcadores genéticos de mala respuesta, siendo las traslocaciones, las alteraciones más frecuentes. La leucemia del anciano tiene peor pronóstico y existe controversia con respecto a si debe recibir tratamiento o solamente soporte; es de esperar que tengan menor supervivencia si solo reciben medidas de soporte- También existe discusión si en ancianos se debe utilizar dosis convencionales o reducidas, siendo las últimas mejor toleradas.
Evolución
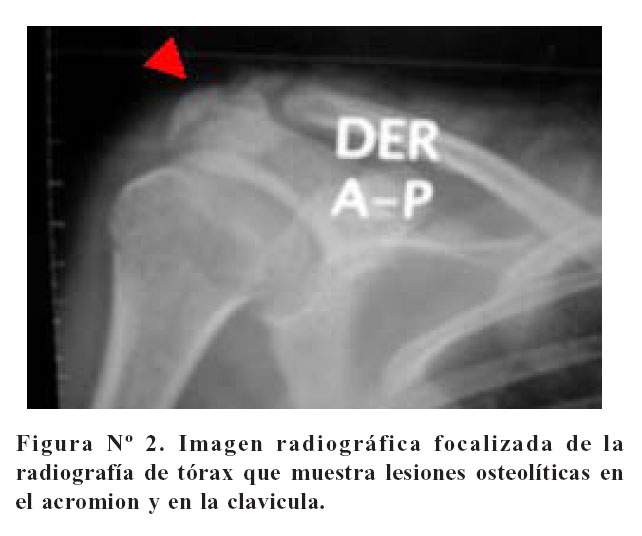
La artralgia de hombro derecho era una de las molestias principales referidas por la paciente. Al ver la placa de tórax, se visualiza el hombro derecho que presenta lesiones líticas en el acromion y en la cabeza del húmero ( Figura Nº2).
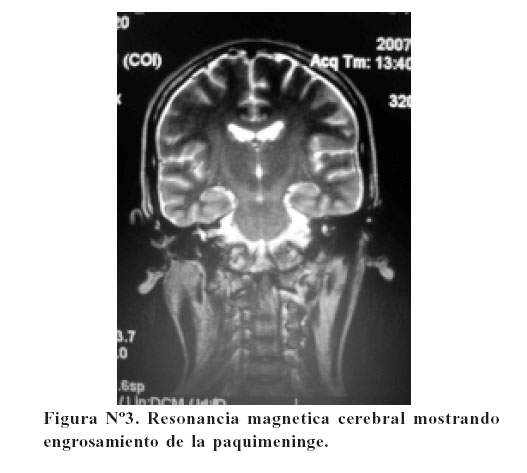
La paciente fue transferida al INEN donde recibió quimioterapia; al 4to día de tratamiento presentó convulsiones, el EEG fue normal, la TAC cerebral no mostró lesión aparentemente y la RM cerebral mostró engrosamiento de la paquimeninge ( Figura Nº3).
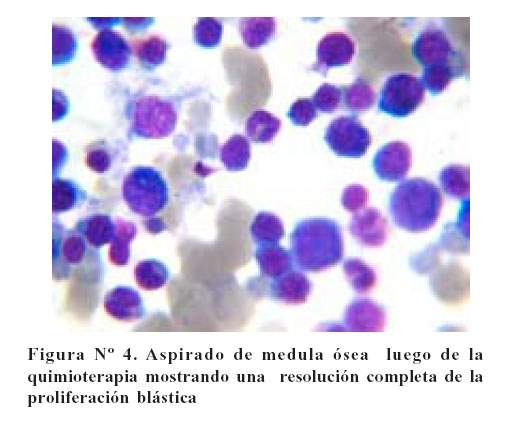
Posterior a la quimioterapia, el estudio de médula ósea mostró celularidad adecuada para la edad, mayor cantidad de tejido adiposo, aspecto celular heterogéneo y a mayor acercamiento se observaron las 3 series hematopoyéticas presentes, reportándose <1% de blastos. Salió de alta en condición de remisión completa ( Figura Nº4).
Dr. Cipriani: ¿Se le pudo hacer inmunohistoquimica para determinar la estirpe celular?
Dra. Jhong: si se le realizó, fué peroxidasa negativo (sugerente de leucemia de estirpe linfoide); además, se le realizó inmunofenotipo determinando leucemia linfocitica aguda y en el análisis citogenético encuentraron mutaciones que comprometen el cromosoma 7.
REFERENCIAS BIBLIOGRAFICAS
1. Bassil KL. Cancer health effects of pesticides: systematic review. Can Fam Physician 2007; 53 (10): 1704-11. [ Links ]
2. Sanborn N. Non-cancer health effects of pesticides: systematic review and implications for family doctors. Can Fam Physician 2007; 53 (10): 1704-11. [ Links ]
3. Knudson AG. Cancer genetics. Am J Med Genet 2002; 111 (1): 96-102. [ Links ]
4. Olney RK, Aminoff MJ. Weakness, myalgias, disorders of movements, and imbalance. En: Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, Jameson JL, eds. Harrisons principles of internal medicine. 15th ed. New York, NY: McGraw-Hill; 2001.p. 119–127. [ Links ]
5. Butturini A, Gale RP. Causes of leukemia. En: Newlan AC. Editor. Cambridge Medical Reviews: Haematological Oncology. Cambridge University Press; 1991. p.103-117. [ Links ]
6. Lipshultz S. The effect of Dexrazoxane on myocardial Injury in Doxorubicin-treated Children with Acute Lymphoblastic leucemia. N Engl. J. Med 2004; 351:145-53. [ Links ]
7. Hoelzer DR. Diagnosis and treatment of adult lymphocytic leucemia. Neoplastic diseases of the blood.New York: Churchill Livingstone, 1991.p.253-274. [ Links ]
8. Jabbour E. Acute tumor lysis syndrome: update on therapy. Rev Med Interne 2005; 26(1):27-32. [ Links ]

