










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Medica Herediana
Print version ISSN 1018-130XOn-line version ISSN 1729-214X
Rev Med Hered vol.21 no.3 Lima July 2010
Diabetes mellitus tipo 2 y resistencia a la insulina.
Type 2 diabetes mellitus and Insulin resistence.
Enrique Cipriani-Thorne1, Alberto Quintanilla2.
1
Profesor Principal de Medicina, Universidad Peruana Cayetano Heredia, Clínica San Felipe. Lima, Perú.2
Residente de cuarto año de Endocrinología, Hospital Arzobispo Loayza, Universidad Peruana Cayetano Heredia. Lima, Perú.
RESUMEN
Se analizó la literatura de los últimos 15 años para poder objetivar los temas que son materia de esta comunicación. En primer lugar se analiza en qué consiste el llamado Síndrome Metabólico, sus características; a continuación se resume la historia natural de la Diabetes Mellitus tipo II y su patogenia. Se revisa la importancia de la resistencia a la insulina en el músculo estriado principal órgano de combustión de la glucosa y el factor mitocondrial de la resistencia a la insulina. Se analiza además la importancia de resistencia a la insulina con respecto al metabolismo del hígado y en particular a las alteraciones del metabolismo del colesterol y de los triglicéridos. Se hace un comentario extenso de cómo así la Diabetes Mellitus Tipo II es considerada en la actualidad como una enfermedad de origen inflamatorio y por último se discuten las consecuencias de la hiperinsulinemia, dado que el exceso de insulina es producido por el bloqueo en la línea metabólica de su acción más no en su línea de producir proliferación tisular.(Rev Med Hered 2010;21:160-170).
PALABRAS CLAVE: Resistencia insulina, diabetes mellitus.
SUMMARY
The literature of the last 15 years was reviewed to document the facts of this paper. In this place the characteristics of the metabolic syndrome are summarized along with the natural history of Diabetes Mellitus II. The importance of insulin resistance in striated muscle is detailed as well as reviewed the mitochondrial factor. Currently Diabetes Mellitus II is taken to be an inflammatory disease, this issue is also reviewed. And finally we discuss the role of excess insulin in generating the late complications of Diabetes Mellitus II. (Rev Med Hered 2010;21:160-170).
KEY WORDS: Insulin resistance, Diabetes Mellitus.
INTRODUCCIÓN
La resistencia a la insulina asociada a factores de susceptibilidad genética condicionan una serie de alteraciones clínicas: hipertensión arterial, intolerancia a la glucosa que en última instancia deriva en diabetes mellitus tipo 2, arterioesclerosis como consecuencia de la disminución del colesterol HDL, la elevación del LDL y de los triglicéridos (1) (Figura 1).
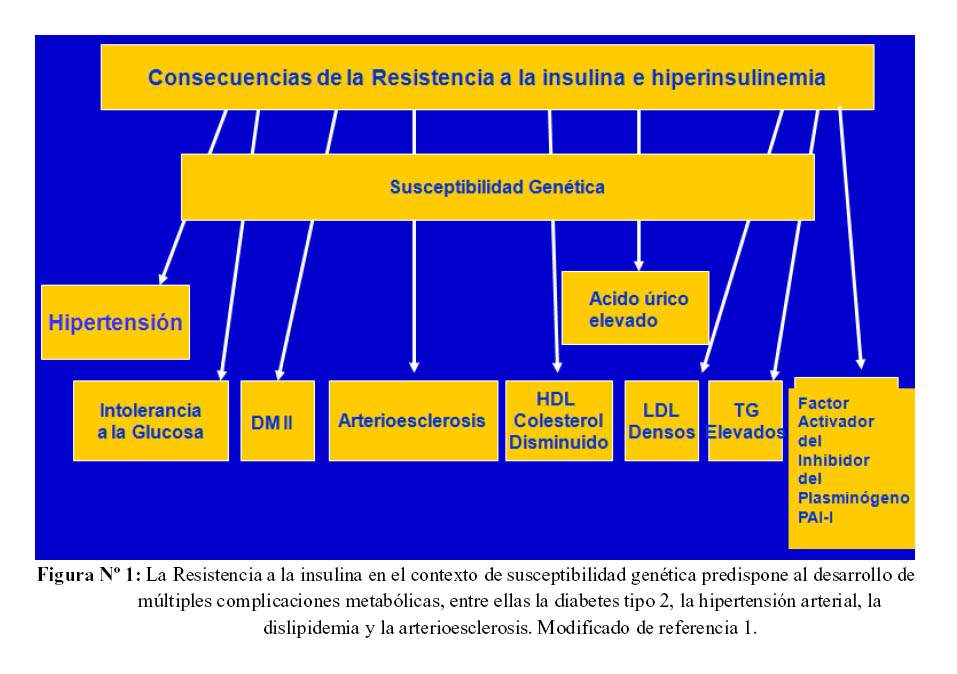
Síndrome Metabólico
La resistencia a la insulina es el componente central del llamado síndrome metabólico descrito por Reaven en 1988, los componentes de este síndrome giran alrededor de este fenómeno, en la actualidad los criterios de diagnostico incluyen la presencia de por lo menos tres de los siguientes criterios (2):
1. Obesidad: definida como perímetro abdominal igual o mayor de 102 cm en varones y 88 cm en mujeres.
2. Dislipidemia: caracterizada por una cifra igual o mayor de 150 mg de triglicéridos/dL.
3. Niveles de colesterol HDL menores de 40 mg/dL en varones o de 50 mg/dL en mujeres.
4. Presión arterial sistólica mayor o igual de 130 mm Hg y diastólica mayor o igual de 85 mm Hg.
5. Niveles de glucosa en sangre mayores a 110 mg/dL.
Existen otros factores de riesgo asociados con el síndrome metabólico y que parecen depender de la resistencia a la insulina y a la consiguiente hiperinsulinemia, tales como: el incremento en la proteína C reactiva, alteraciones del fibrinógeno, exceso en la circulación de citoquinas como la interleuquina 6 (IL 6), el factor de necrosis tumoral alfa (TNF), moléculas de adhesión intracelular y de células vasculares (ICAM – 1 y VCAM), el factor VIII y de factores protromboticos tales como el inhibidor del activador de plasminogeno (PAI 1), el dimero D y el fibrinopeptido A, además de la presencia de "stress" oxidativo, como es el caso del colesterol LDL oxidado y otros lípidos peroxidados (3,4).
La resistencia a la insulina es medida con diversos métodos de laboratorio, ninguno de ellos es sensible. El diagnostico de esta suele deducirse cuando se encuentren los hallazgos clínicos o de laboratorio que caracterizan a este llamado síndrome metabólico
La diabetes mellitus tipo 2 (DM2) es la punta del iceberg, que tiene un primer estadio, algunas de las alteraciones ya mencionadas, la curva de tolerancia a la glucosa (CTG) aun es normal, en presencia de hiperinsulinemia y los individuos muchas veces tienen obesidad, con incremento en la relación cintura – cadera; en esta etapa, se inicia la aceleración del envejecimiento arterial (aterogénesis) y el incremento en la prevalencia de hipertensión arterial. En un segundo estadio, ocurre alteración en la tolerancia a la glucosa, con niveles post prandiales de glucosa elevados, la producción hepática de glucosa se incrementa y el transporte de glucosa al interior de las células disminuye, pues empieza a ocurrir déficit en la secreción de insulina. En un tercer estadio, la diabetes mellitus se hace clínicamente detectable y se incrementa la presencia de macroangiopatia y microangiopatia que ya ocurren en individuos con CTG alterada, para aumentar en proporciones desmesuradas cuando la DM está mal controlada (5) (Figura 2).
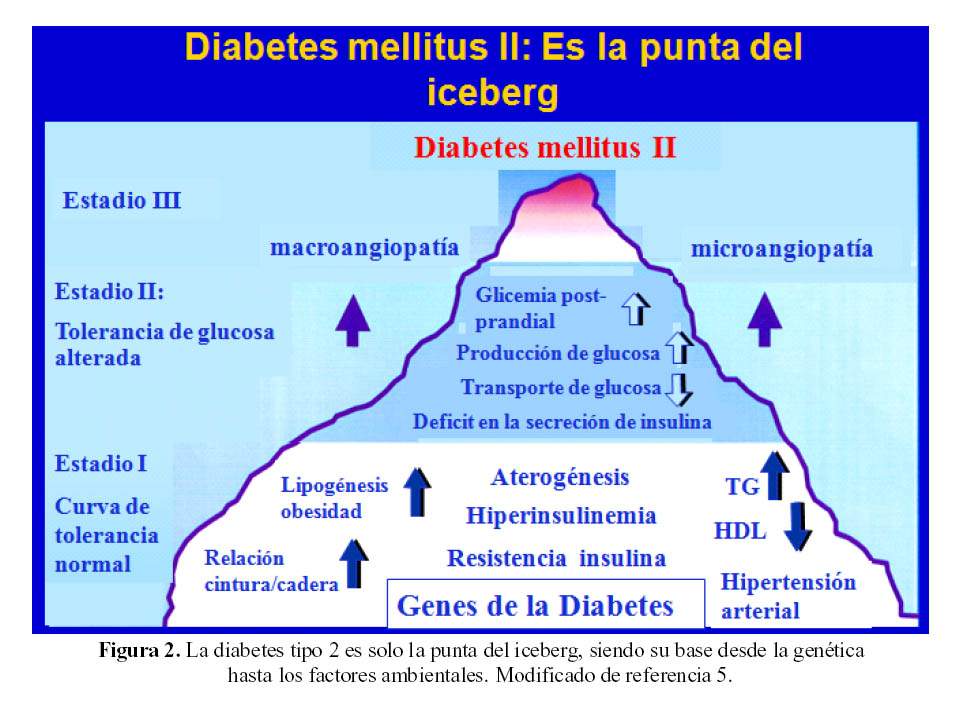
Historia Natural de la Diabetes Mellitus Tipo 2
La DM2 es una enfermedad de presentación muy diversa, con alteraciones genéticas que definen la edad de su aparición clínica y la importancia relativa de sus alteraciones en relación con factores ambientales (alimentación y obesidad). Los casos de DM2 con alteración monogénica son raros y se presentan desde el nacimiento hasta la adolescencia; en ellos el factor obesidad es secundario (6). En las formas del adulto la influencia del medio ambiente cobra mucha mayor importancia (Figura 3).

La historia natural de la DM2 del adulto se caracteriza por ser poligénica, con una progresiva disminución de la secreción de la insulina asociada a la alteración paulatina del control de la glicemia; la alteración de la enfermedad es gradual (6) (Figura 4).
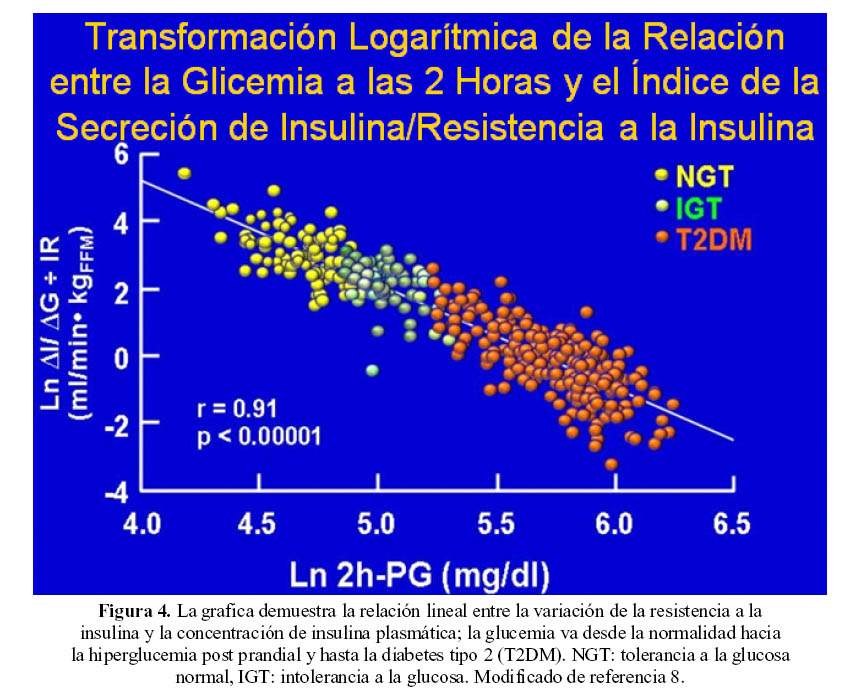
En el metabolismo de la glucosa mediado por insulina durante la CTG, la cifra media de las glicemias se desplazan hacia arriba en sujetos obesos con CTG alterada y paralelamente se elevan los niveles de insulina conforme evoluciona la enfermedad, la glicemia aumenta mas y caen los niveles de insulina en simultaneo con el decrecimiento en la captación tisular de glucosa (7).
Patogenia de la Diabetes Mellitus tipo 2
En una versión actualizada de la patogenia de la DM2 De Fronzo ha identificado hasta ocho mecanismos a los que denomina "el octeto del mal agüero" (8). Todos ellos condicionan hiperglicemia:
1. Disminución del efecto de incretinas.
2. Incremento de la lipolisis.
3. Incremento en la reabsorción tubular de glucosa en el riñón.
4. Disminución de la captación de glucosa por el musculo.
5. Disfunción en los neurotransmisores cerebrales.
6. Incremento de la gluconeogénesis por el hígado.
7. Incremento en la secreción de glucagon por las células alfa del páncreas.
8. Disminución paulatina en la secreción de insulina por el páncreas.
Resistencia a la insulina y músculo estriado
El músculo estriado es el tejido que en condiciones normales, consume la mayor proporción de glucosa en el organismo hasta el 85%
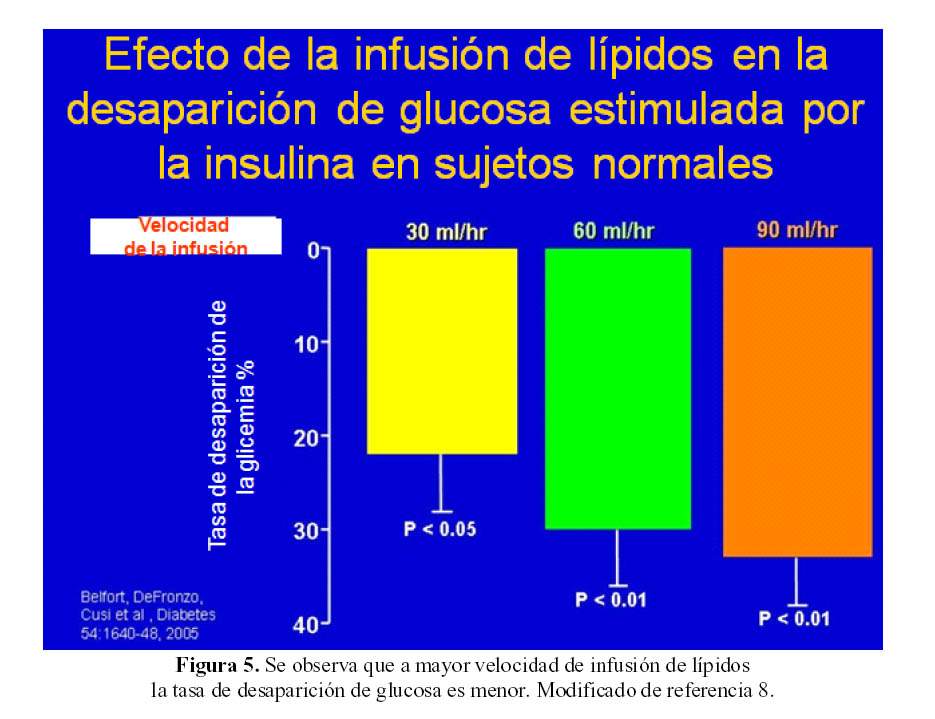
Estudiando el efecto de la infusión de lípidos sobre la desaparición de glucosa sanguínea estimulada por la insulina en sujetos normales, se observó que conforme se incrementaba la velocidad de infusión de lípidos, ocurre una menor caída de la glicemia (Figura 5) lo que permite afirmar que los ácidos grasos disminuyen de manera importante el metabolismo de glucosa en el músculo (7).
El incremento de la lipolisis en el tejido adiposo, moviliza ácidos grasos hacia la circulación y al entrar al músculo, se incrementa la oxidación de estos, con una simultánea disminución en la utilización de glucosa por el músculo. El incremento de movilización de ácidos grasos, también repercute en el metabolismo hepático, pues al oxidarse en este órgano se aumenta la gluconeogénesis (9).
Se ha descrito una alteración en la distribución de fibras musculares esqueléticas en pacientes con DM2 consistente en: a) una disminución del 16% de las fibras musculares estriadas tipo 1; estas fibras se caracterizan por un metabolismo oxidativo elevado y son las que confieren la capacidad de resistencia en la actividad muscular; y b) un incremento del 49% en las fibras con metabolismo glicolitico rápido, que tienen una eficacia metabólica mucho menor (10). En el tejido muscular la actividad de la ATPasa se correlaciona de manera inversa con los niveles de hemoglobina glicosilada, a mayor nivel de hemoglobina glicosilada menor actividad de ATPasa; además, se describe una correlación directa entre la actividad ATPasa muscular y la sensibilidad a la insulina; así como una correlación directa con la eficacia de la contracción muscular (10).
Factor mitocondrial
Las mitocondrias son el embudo metabólico de las células, son las organelas donde se produce la energía de todo el organismo. En ellas ocurre la interconversion entre los diferentes sustratos: aminoácidos, glucosa, glicerol y ácidos grasos en el ciclo de Krebs. La actividad mitocondrial genera energía en la forma de ATP; para esto en condiciones normales, el consumo de oxigeno esta acoplado íntimamente a la generación de ATP y a la producción de agua (Figura 6).
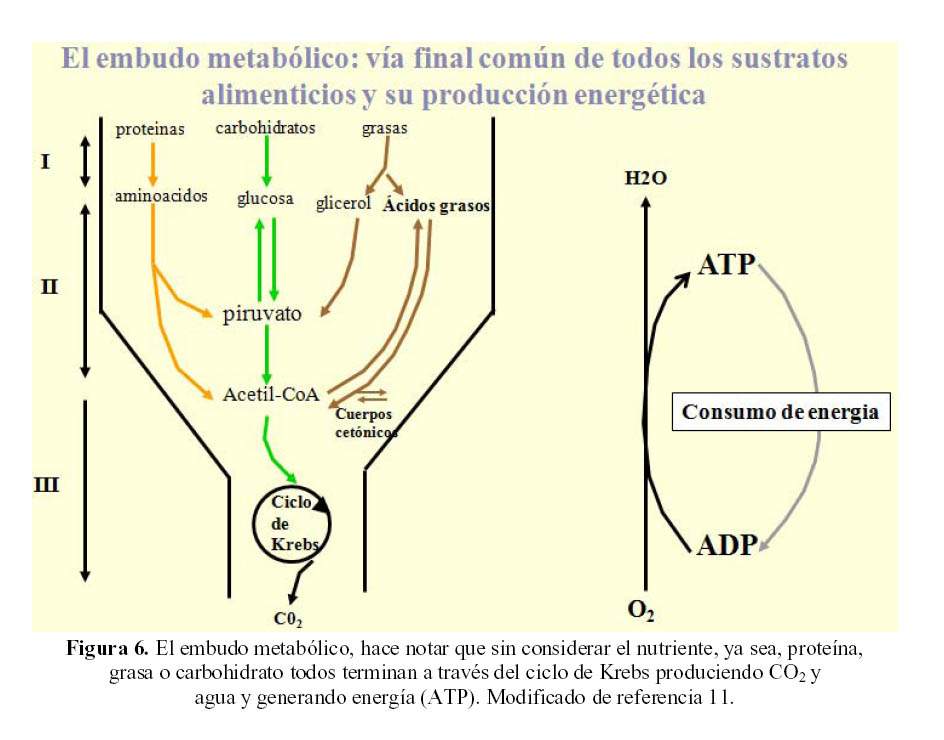
En casi todas las células del organismo, con excepción de las neuronas, hematíes y células de la médula renal, el transporte de glucosa estimulado por insulina, requiere de la migración de transportadores específicos hacia la membrana celular; en el caso del músculo estriado, tejido adiposo, miocardio, hepatocito, etc, son los denominados GLUT4 (12).
La unión de la insulina con su receptor de membrana activa a una cadena de proteínas: los sustratos del receptor de insulina (IRS), que son fosforilados en sus residuos de tirosina iniciando un proceso autocatalitico que culmina con la activación del sistema de los fosfoinositoles que producen los segundos mensajeros para llevar GLUT4 a la membrana celular.
Un mecanismo importante para la resistencia a la insulina, es el fenómeno de disfunción mitocondrial; el exceso de ácidos grasos en la circulación, asociado a la reducción en el número de mitocondrias, produce un incremento en el nivel de ácidos grasos intracelulares y también del diacilglicerol. Estas moléculas activan la proteína kinasa C (PKC) que a su vez activa la cascada de la serina kinasa, llevando al incremento en la fosforilacion de los residuos de serina en el IRS1. EL incremento en la fosforilacion de serina del receptor IRS impide la fosforilacion de residuos de tirosina en IRS1; lo cual a su vez inhibe la actividad de la fosfoinositol kinasa, determinando en última instancia la supresión del transporte de glucosa inducida por insulina (13).
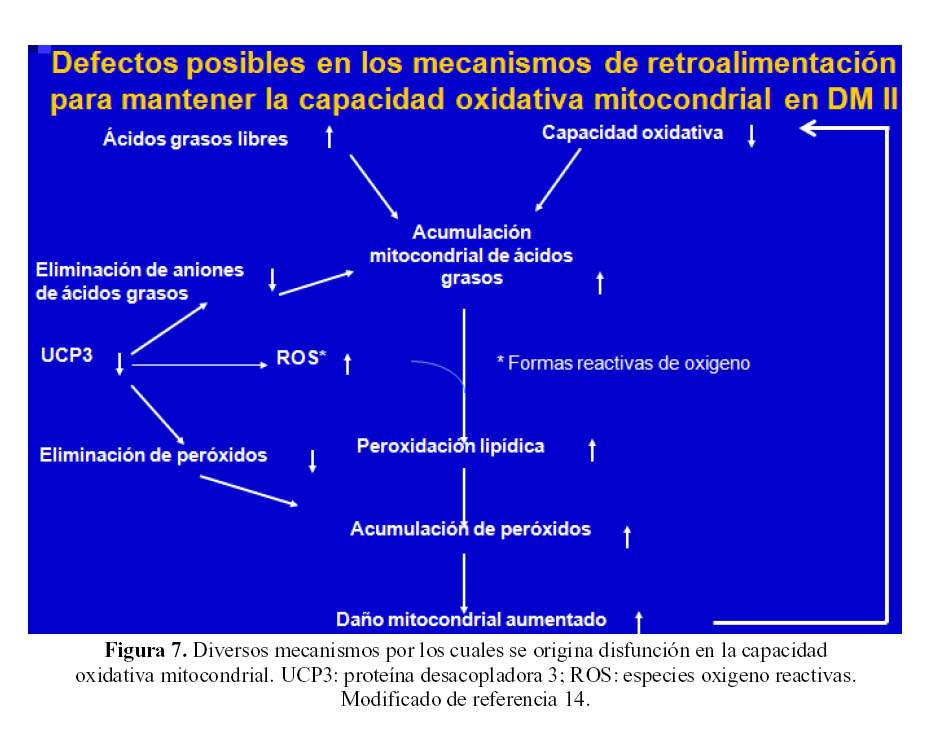
Existen varios defectos en los mecanismos de retroalimentación para mantener la capacidad oxidativa mitocondrial en DM2 (Figura 7): a) la elevación de los ácidos grasos libres lleva a la acumulación mitocondrial de ácidos grasos; contribuye a esto, b) la disminución en la función de la proteína desacopladora tipo 3 (UCP3) que condiciona disminución en la eliminación de aniones de ácidos grasos, c) la disminución de la actividad de UCP3 incrementa la producción de formas reactivas de oxigeno que aumentan la peroxidacion lipidica, d) lo cual redunda en incremento de el daño mitocondrial, que conlleva e) una disminución en la capacidad oxidativa celular (14).
Resistencia a la Insulina, Hígado y Dislipidemia
La movilización anormal de ácidos grasos originados en el tejido adiposo, provoca una sobrecarga en la función hepática, agravada por el exceso de producción de citoquinas en el hígado, que ocurre por la acumulación de grasa en este órgano, condicionando la llamada esteato hepatitis no alcohólica (14).
El exceso de ácidos grasos que afluyen al hígado causa un incremento en la síntesis de VLDL y como resultado (Figura 8) del catabolismo del VLDL "grandes", se producen en última instancia LDL "densos". Los LDL "densos" tienen una mejor capacidad de infiltrar la pared arterial que los LDL menos "densos" y también dan productos de peroxidacion (ROS) (15).
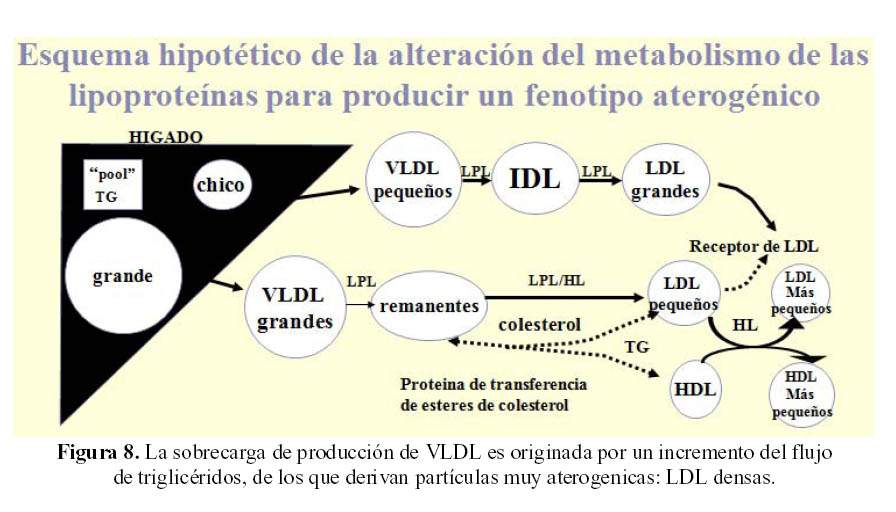
Existe una correlación directa entre resistencia a la insulina y los LDL "densos" de menor tamaño; la separación de los LDL "densos" del LDL menos "densos" es gradual. Así como hay una variedad de HDL, unos que son más grandes que los otros, no hay una línea divisoria clara entre los LDL y los HDL, ni dentro de estas mismas formas moleculares del transporte del colesterol en la sangre. La separación de estas moléculas se hace por métodos de experimentación que son costosos y aun no tiene llegada al laboratorio clínico (15).
Factor inflamatorio
El factor inflamatorio que ocurre a consecuencia del exceso en tejido adiposo, ha cobrado particular importancia en la comprensión de los problemas de la DM2 (16).
El exceso de tejido graso produce una resistencia a la insulina por el incremento en la lipolisis, que libera ácidos grasos a la circulación; además en este tejido ocurre paralelamente un aumento en la infiltración por macrófagos, que tienen origen común con los pre – adipocitos; la infiltración es agravada por la producción de citoquinas inflamatorias; asociado a esto se incrementa la producción de formas reactivas de oxigeno (16).
Durante el proceso inflamatorio en el tejido adiposo, los adipocitos secretan pequeñas cantidades de TNF que estimula a los pre adipocitos a producir MCP 1 (proteína quimioatrayente de monocitos) que atrae el ingreso de monocitos, el mismo fenómeno es generado por células endoteliales (16).
En la obesidad el incremento en el tamaño de los adipocitos genera daño por "stress oxidativo" desencadenado por el exceso de lipolisis. Todos estos mecanismos terminan produciendo citoquinas inflamatorias con la siguiente alteración de la función del tejido adiposo y la resistencia a la insulina en este tejido (16).
Conforme la persona incrementa peso y aumenta la producción de IL6, IL1, TNF se dan modificaciones genéticas en el factor NF kB y el JNK (16).
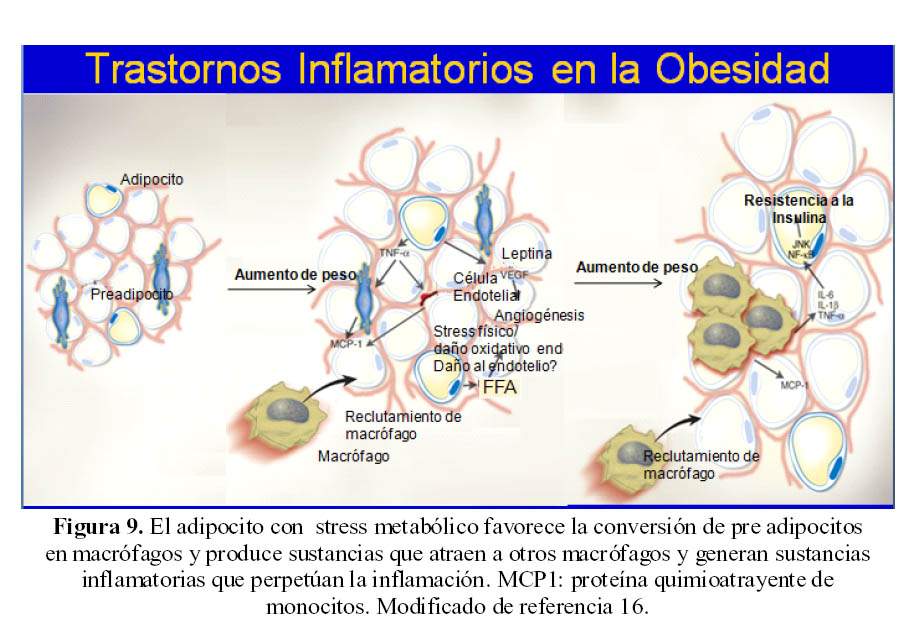
En condicionen normales los adipocitos almacenan grasa: su rol en la homeostasis metabólica; en tanto que los macrófagos trabajan en la respuesta inflamatoria, aunque ambos tipos celulares tienen la capacidad para realizar esas funciones. En la obesidad ese equilibrio se trastorna de macrófagos y la generación de citoquinas inflamatorias generadas por los propios adipocitos (16).
Los macrófagos tienen una importancia primaria en la respuesta inflamatoria y los adipocitos en la respuesta metabolica; sin embargo existe una interconversion de pre – adipocitos a macrófagos y hay funciones que se sobreponen para ambas células (16) (Figura 9).
La inflamación del tejido adiposo es un paso importante en el desarrollo de resistencia a la insulina además de favorecer el desarrollo de la arterioesclerosis por la dislipidemia y la acumulación lípidos en los macrófagos los que se convierten en las células espumosas en la pared del sistema vascular.
Los adipocitos y los macrófagos comparten muchas características tales como la expresión de citoquinas, proteínas ligadoras de ácidos grasos (FABP), receptores de hormonas nucleares, etc. Las vías de activación del PPAR ã y LXR se oponen al fenómeno inflamatorio y promueven la salida del colesterol de los macrófagos y favorecen el depósito de lípidos en los adipocitos.
Las vías inflamatorias pueden ser iniciadas por mediadores extracelulares como son las citoquinas y lípidos, o por mecanismos intracelulares tales como el "stress" del retículo endoplasmático o el exceso de producción de especies reactivas de oxigeno en las mitocondrias.
La información de los mediadores señalados convergen en rutas pro inflamatorias que incluyen a las kinasas JNK (Jun N terminal Kinasa) e IKK (I kappa B Kinasa). Estas rutas llevan a la producción de otros mediadores inflamatorios a través de los reguladores de transcripción así como la inhibición directa de las vías de acción de la insulina (16).
Otras rutas como las mediadas a través de las proteínas SOCS (Supresor de señalización de citoquinas) e iNOS (Sintasa de oxido nitrico) también están comprometidas en la inhibición inflamatoria de la acción de la insulina (16).
El control de la inflamación a un nivel más proximal esta dado por las FABP que probablemente secuestran ligandos de factores de transcripción, antiinflamatorios, la ausencia de FABP limita la inflamación.
En condiciones de sobrenutrición la célula adiposa debe tener un balance entre metabolismo e inflamación; este es un reto importante; los mismos procesos que se requieren para utilizar nutrientes tales como el metabolismo oxidativo en la mitocondria o el incremento de la síntesis proteica en el retículo endoplásmico pueden inducir una respuesta inflamatoria (16).
La activación del factor nuclear kappa B (NF kB) es el eje del que dependen los mecanismos de acción inflamatoria y también sobre el cual inciden los diferentes mecanismos inflamatorios tanto naturales del organismo, así como los medicamentos que usamos como agentes inflamatorios (17) (Figura 10).
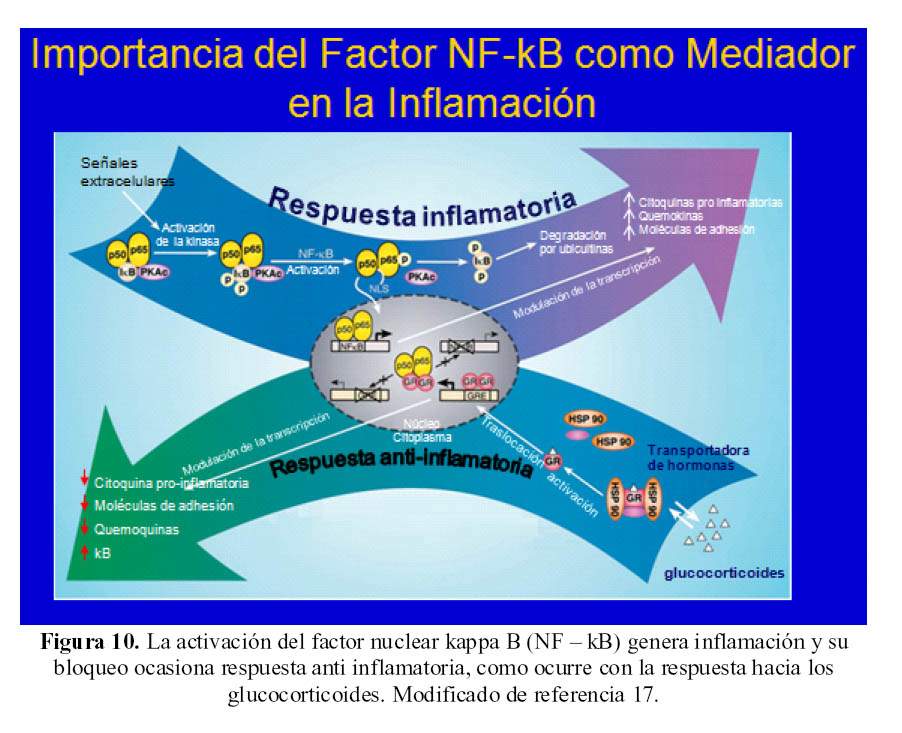
La activación de este factor tiene la siguiente secuencia: los heterodimeros inactivos del NF kB se retienen en el citoplasma por una unidad inhibitoria de este NF kB que se llama IkB. El NF kB del citoplasma está ligado a una subunidad catalítica de una proteína kinasa A (PKA). Como respuesta a una señal de activación la kinasa IkB fosforila a IkB tornándose asequible a su degradación por ubiquitinación; mientras que la PKA fosforila a la subunidad p65 del NF kB.
Este proceso de activación expone a una señal de localización en el núcleo celular permitiendo que el heterodimero de NF kB migre al interior del núcleo donde interactúa con elementos de transcripción genética (17).
Glucagon y DM2
En la diabetes tipo 2 hay un incremento en la gluconeogénesis hepática acoplado a la disminución de la síntesis de glucógeno y también al incremento de la glucogenolisis. Estos fenómenos se deben tanto a la reducción de la actividad insulinica como al incremento relativo en la concentración de glucagon plasmático.
Para entender la importancia de las acciones hormonales sobre el hígado es necesario recordar que el drenaje venoso del páncreas va directamente a la circulación portal; además el hígado recibe una concentración de insulina y glucagon mucho mayores que las existentes en la circulación periférica (18).
Durante la curva de tolerancia a la glucosa en pacientes con DM2 la secreción de insulina disminuye a pesar del incremento en los niveles de glucosa, en tanto que los niveles de glucagon persisten sin tener alteración (18).
Hiperinsulinemia – Consecuencias
La insulina puede ser considerada como la hormona responsable del metabolismo de los carbohidratos, particularmente de la glucosa. Pero es necesario recalcar su importancia fisiológica como hormona anabólica, así como sus efectos dañinos cuando está crónicamente elevada.
El exceso de la insulina en sangre favorece el estimulo de acciones celulares que no se encuentran bloqueadas en la resistencia a la insulina. La resistencia a la insulina bloquea las rutas del metabolismo del transporte de glucosa que conllevan en última instancia a la síntesis de glucógeno y de lípidos; pero no se bloquea la ruta de la proteína kinasa activadora de mitogenos (MAPK) que conlleva a la proliferación de musculatura vascular, producción de moléculas de adhesión celular, disminución de la síntesis de oxido nitroso vascular (19).
La poliquistosis ovárica está relacionada con la resistencia a la insulina; hasta un 8% de mujeres la padecen tanto con peso normal como con sobrepeso u obesidad. En este síndrome existe como factor común la resistencia a la insulina y parece tener un rol significativo en su desarrollo. En la polquistosis ovárica hay alteraciones y cuando estos cambios son revertidos por drogas que sensibilizan a los tejidos a la insulina se normaliza la fisiología ovárica y se revierten con bastante éxito los daños que se observan en la poliquistosis ovárica entre ellos se mejora o regularizan los ciclos menstruales.
El exceso de insulina estimula acciones celulares que no se encuentran bloqueadas en la resistencia a la insulina; la activación de la MAPK tiene como consecuencia disfunción endotelial, arterioesclerosis, enfermedad coronaria e hipertensión arterial (19).
El análisis de las alteraciones de las células beta en la DM2, la función de los PPAR, el coactivador del receptor PPAR (PGC – 1), el factor incretina y el tratamiento de la DM2 en el contexto de estos conocimientos serán motivo de una próxima publicación.
REFERENCIAS BIBLIOGRÁFICAS
1. Bouchard C. Genetics and the metabolic syndrome. Int J Obes Relat Metab Disord 1995;12 (S1):S52–S59. [ Links ]
2. Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults. Executive Summary of Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adults Treatment Panel III). JAMA 2001; 285: 2486-2497. [ Links ]
3. Creager MA, Luscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease Pathophysiology, clinical consequences, and medical therapy: part I. Circulation 2003; 108: 1527–32. [ Links ]
4. Ceriello A, Motz E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler Thromb Vasc Biol 2004; 24: 816–23. [ Links ]
5. Matthaei S, Stumvoll M, Kellerer M, Hans-Ulrich Häring HU. Pathophysiology and Pharmacological Treatment of Insulin Resistance. Endocr Rev 2000; 21: 585-618. [ Links ]
6. Vaxillaire M, Froguel P. Monogenic Diabetes in the young, pharmacogenetics and relevance to multifactorial forms of Type 2 Diabetes. Endocr Rev 2008; 29(3):254–264. [ Links ]
7. De Fronzo R. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1988; 37: 667-687. [ Links ]
8. DeFronzo R. Banting Memorial Lecture. San Francisco: American Diabetes Association 68th Scientific Sessions; 2008. [ Links ]
9. DeFronzo R. Pathogenesis of type 2 diabetes: metabolic and molecular implications of identifying diabetes genes. Diabetes Rev 1997; 5:177–269. [ Links ]
10. Oberbach A. Altered Fiber distribution and fiber-specific glycolytic and oxidative enzyme activity in skeletal muscle of patients with Type 2 Diabetes. Diabetes Care 2006; 29: 895- 900. [ Links ]
11. Flatt JT. Substrate Utilization and Obesity. Diabetes Reviews 1996; 4: 433–449. [ Links ]
12. Haring HU Pathogenesis of type II diabetes: are there common causes for insulin resistance and secretion failure? Exp Clin Endocrinol Diabetes 1999; 107 (S2): S17–S23. [ Links ]
13. Bradford BL, Shulman GI. Mitochondrial Dysfunction and Type 2 Diabetes. Science 2005; 307 (5708): 384 –387. [ Links ]
14. Schrauwen P. Oxidative capacity, lipotoxicity, and mitochondrial damage in Type 2 Diabetes. Diabetes 2004; 53: 1415. [ Links ]
15. Wellen KE. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest 2003; 112:1785-1788. [ Links ]
16. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J. Clin Invest 2005; 115:1111–1119. [ Links ]
17. McKay LI, Cidlowski JA. Molecular Control of Inmune/Inflamatory Responses. Interactions between Nuclear Factor kB and Steroid Receptor Signaling Pathways. Endocrine Reviews 1999; 20(4): 435 – 459. [ Links ]
18. Butler pc, Rizza RA. Contribution to postprandial hyperglycemia and effect on initial splanchnic glucose clearance of hepatic glucose cycling in glucose- intolerant or NIDDM patients. Diabetes 1991; 40(1): 73– 81. [ Links ]
19. Ranganath M. Cardiovascular actions of insulin. Endocr Rev 1997; 28(5):463–491. [ Links ]

