










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Medica Herediana
versión impresa ISSN 1018-130X
Rev Med Hered v.22 n.1 Lima ene. 2011
Síndrome urémico hemolítico atípico de presentación neonatal.
Atypical hemolytic uremic syndrome of neonatal presentation.
Jenny Ponce1, César Salinas2, Reyner Loza1.
1
Departamento de Pediatría, Unidad de Nefrología pediátrica, Hospital Nacional Cayetano Heredia. Lima, Perú.2
Departamento de Patología. Clínica Médica Cayetano Heredia. Lima, Perú.
RESUMEN
El Síndrome urémico hemolítico (SUH) tiene formas típicas y atípicas. Se describe una variedad de formas genéticas con pobre pronóstico. Presentamos un bebé prematuro de 36 semanas, de bajo peso al nacer, quien a las 2 semanas de vida cursó con sepsis y necrosis intestinal siendo sometido a cirugía para realizarle ileostomía. Evolucionó con hipertensión arterial, hematuria, falla renal aguda y proteinuria persistente. A los 2 meses de vida, posterior al cierre de ileostomía, cursó con shock séptico y falleció. La biopsia renal post mortem mostró cuadro compatible de SUH. Dos años después, un hermano debutó a los 2 días de vida con síndrome nefrótico congénito. El estudio genético reveló que la madre era portadora del gen NPHS1 y el padre, del Factor I de complemento. El segundo hijo era portador de ambos genes. (Rev Med Hered 2011;22:29-33).
PALABRAS CLAVE: SUH atípico, gen del factor I, falla renal aguda.
SUMMARY
Hemolytic Uremic Syndrome (HUS) has typical and atypical presentations. A variety of genetic forms, with poor prognosis are described. We report a 36 week premature baby, low birth weight, who at 2 weeks of life evolved with sepsis and intestinal necrosis undergoing surgery for ileostomy, hypertension, hematuria, acute renal failure and persistent proteinuria. At 2 months, after ileostomy closure, developed irreversible septic shock and died. Post-mortem renal biopsy was compatible with HUS. Two years later, a brother presented after 2 days of birth with congenital nephrotic syndrome. Genetic studies revealed that the mother was carrying the gene NPHS1 and the father, factor I of complement. The second child was a carrier of both genes. (Rev Med Hered 2011;22:29-33).
KEY WORDS: Atypical HUS, factor I gen, acute kidney failure.
INTRODUCCIÓN
El síndrome urémico hemolítico (SUH) se caracteriza por la triada anemia hemolítica, trombocitopenia y falla renal aguda (1-7). La presentación típica es la más común y se presenta con pródromos de diarrea; en 90% de casos y tiene buen pronóstico (1). La forma atípica en el 10% restante, tiene pobre pronóstico y alta mortalidad, más del 25% en la fase aguda y requiriendo terapia dialítica, en el 50% de casos (2,6,7).
Se ha descrito una variedad de formas genéticas, 50% con alteración en los genes que regulan el complemento (factor H, factor I, MCP, factor B y C3) y 10% por la presencia de anticuerpos contra el factor H (3-6).
El factor I es una proteasa de serina que regula la ruta alterna del complemento, al escindir la cadena alfa del C3b en presencia de proteínas cofactoras (3,4,7,8). Se ha reportado menos de 20 mutaciones (3,9), siendo descritas en su mayoría como heterocigotos (6,7,10-12). La mayoría de éstas induce a ausencia de síntesis de proteínas y solo unas pocas, a deficiencia funcional, por lo que se puede encontrar concentraciones normales o reducidas del factor I, así como una reducción variable del C3 (3,5,8). Esta última mutación es causa del 3 al 12% de casos de SHU atípico, manifestándose por primera vez antes de los 3 meses de edad, en promedio 2 meses (3,6,7,13). El cuadro clínico es similar a la deficiencia hereditaria de C3, siendo la característica más común la presencia de infecciones piógenas recurrentes, que se inician usualmente en la infancia temprana (9,14). La evolución de la enfermedad es variable, la mitad se recupera y la otra mitad muere o evoluciona rápidamente a enfermedad renal crónica terminal (13).
Debido a la poca frecuencia, se describe un caso de SUH atípico de presentación neonatal posiblemente debido a mutación del gen del Factor I del complemento.
Caso clínico
Prematuro varón de 36 semanas, de padres sin antecedentes de importancia. Pesó al nacer 2 440 gramos y su APGAR fue adecuado. Tuvo hematoma en la vena del cordón umbilical y presentó una placenta calcificada. La madre tuvo amenaza de parto pretérmino, recibiendo nifedipino y betametasona como tratamiento. El bebé cursó con distrés respiratorio en las primeras horas de vida, evidenciándose neumotórax derecho y neumomediastino a las 15 horas de nacido, con evolución favorable. Fue dado de alta a los 4 días de vida.
A los 13 días presentó deposiciones con sangre, hipoactividad y pobre succión. Al examen físico presentaba taquipnea y saturación en 96% por oximetría digital, lucía pálido, edematoso, con hipoactividad e hipotonía generalizada. El abdomen lucía distendido con presencia de masa palpable dolorosa en hemiabdomen derecho; tenía residuo bilioso.
Los exámenes de laboratorio mostraron leucocitosis con desviación izquierda, plaquetopenia, acidosis metabólica, hipoglicemia, hipoalbuminemia severa en 1,6 mg/dL, dímero D incrementado, urocultivo positivo a Klebsiella sp y la radiografía de abdomen simple mostró niveles hidroaéreos y neumatosis intestinal. El paciente fue hospitalizado con el diagnóstico de sepsis neonatal tardía y sospecha de enterocolitis necrotizante. Fue sometido a cirugía, realizándosele ileostomía al evidenciarse necrosis intestinal de 30 cm de extensión a nivel de íleo distal. El estudio anátomo patológico mostró necrosis transmural con presencia de exudado fibrinoleucocitario y hemorragia.
El paciente evolucionó con hipertensión arterial e insuficiencia renal no oligúrica. La creatinina se elevó hasta 2,2 mg/dL, la urea hasta 78 mg/dL y la fracción excretada de sodio fue 2,3% evidenciando insuficiencia renal agua (IRA) establecida. En el sedimento urinario se encontró hematuria, proteinuria (3+) y glucosuria. Se observó descenso progresivo de los niveles de hemoglobina y persistencia de plaquetopenia que mejoraron con la resolución de la infección ( Gráfico 1). La creatinina descendió hasta 0,6 mg/dl.
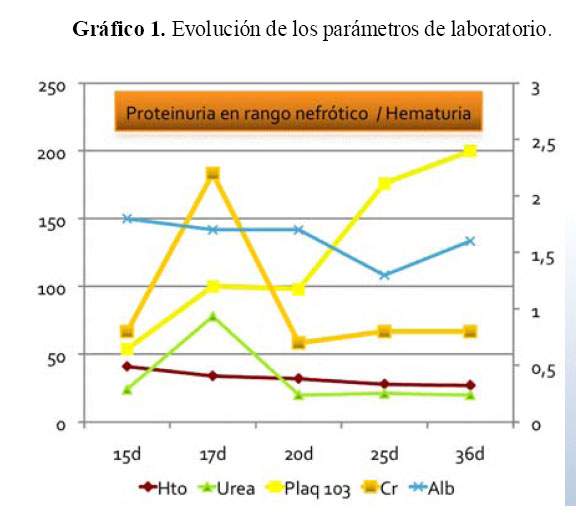
A los 52 días de vida se le realizó cierre de la ileostomía pero nuevamente se observó compromiso de la función renal con recuperación posterior y persistencia de la proteinuria en rango nefrótico, hematuria, glucosuria e hipoalbuminemia. A los 67 días de edad, presentó nuevamente leucocitosis, proteína C reactiva elevada, anemia y plaquetopenia, desarrollando shock séptico con falla renal que requirió diálisis peritoneal aguda, teniendo al final un desenlace fatal. Los cultivos de la punta de catéter venoso central fueron positivos a Klebsiella sp y Estafilococo aureus.
Se extrajo una muestra de riñón por punción post mortem el que mostró edema endotelial con disminución de la luz capilar a nivel del glomérulo renal y cambios vasculares con hiperplasia concéntrica de la capa muscular arteriolar con edema de las células endoteliales. Cambios compatibles con SHU atípico ( Figura 1 y figura 2 ).
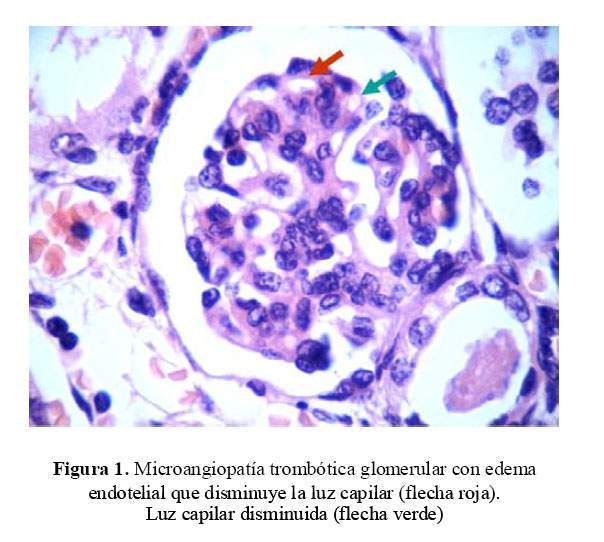
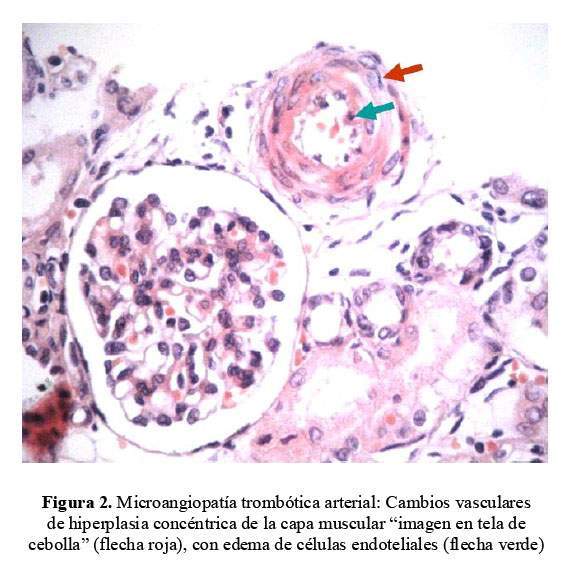
Dos años después, la madre tuvo otro hijo recién nacido (RN) prematuro de 34 semanas, de bajo peso al nacer (2,2 Kg) y placentomegalia con cordón umbilical muy edematoso. No hubo oligohidramnios en las ecografías prenatales. A los 2 días de vida, presentó proteinuria masiva, edema e hipoalbuminemia severa, planteándose el diagnóstico de síndrome nefrótico congénito.
El estudio genético mostró que el RN era portador del gen NPHS1 que codifica la nefrina (estudio realizado por la Dra. Corinne Antignac en Paris) y del gen del factor I del complemento, mutación G261D (estudio realizado por el Dr. Richard Smith - University of Iowa Hospitals and Clinics). Se descartó mutación del factor H, del gen MCD y del gen NPHS2. El estudio en los padres mostró que la madre era portadora de mutación del gen de NPHS1 y el padre, del Factor I.
DISCUSIÓN
El caso presentado es de un paciente con cuadro clínico de SUH atípico posiblemente debido a mutación del gen del factor I del complemento, con debut en los primeros días de vida. La presencia de esta mutación incrementa el riesgo de infecciones principalmente piógenas y de trombosis microvascular (9,14). Nuestro paciente presentó sepsis neonatal con necrosis intestinal; el proceso infeccioso, que se observa como evento precipitante en el 60% de casos (7,11), fue el gatillo para activar la ruta alterna del complemento, produciéndose en forma constante e indiscriminada el depósito de C3b tanto en la pared del microorganismo invasor para conseguir su destrucción, como en el endotelio de las células del huésped, produciéndose daño endotelial. Esta amplificación del daño no fue frenada debido a la presencia de la mutación de una de las proteínas reguladores del complemento, el factor I.
El diagnóstico final fue confirmado por estudios anátomo patológico y genético en la familia, ya que no se pudo realizar el estudio genético en el paciente. La anormalidad patológica predominante se encontró en las arteriolas extraglomerulares y arterias interlobulares, a diferencia de la presentación típica en la que se afecta predominantemente los capilares glomerulares, siendo característico en la presencia de trombosis glomerular, dando la apariencia de agrandamiento del glomérulo que sugiere congestión capilar antes que isquemia (2,6,15). Por otro lado, el estudio genético reveló que ambos progenitores eran portadores de diferentes mutaciones, el padre era portador del factor I y la madre, del NPHS1. El hermano menor, quien debutó con síndrome nefrótico congénito, era portador tanto del gen del NPHS1 (nefrina) como del factor I. Esto nos orienta hacia el posible diagnóstico de síndrome urémico hemolítico atípico por probable deficiencia del factor I.
El SHU atípico familiar ocurre entre 25% (registro francés) (13) y 37% de los casos de SHU (registro italiano) (11), siendo más frecuente la ocurrencia en hermanos; sin embargo, en varias familias la enfermedad ocurre en varias generaciones diferentes (3,4). La ausencia de antecedentes familiares no excluye la posibilidad de transmisión genética (4). La penetrancia genética del SUH asociado a mutación del complemento es aproximadamente 50%. Solo la mitad de los miembros de la familia, quienes portan la mutación, manifiestan la enfermedad; esto ha sugerido que el polimorfismo influye en la predisposición de un individuo a la enfermedad y provee una explicación para la penetrancia incompleta de la enfermedad dentro de las familias (3,4,6). Aún así, son necesarios factores externos asociados a la mutación para que se inicie o progrese la enfermedad, como infecciones virales o bacterianas, drogas, ciertas enfermedades sistémicas, etc (4,7,11); es decir, la mutación confiere una predisposición antes que una causa directa de la enfermedad (11). Esto podría explicar porqué el padre y el hermano que son portadores, no han desarrollado la enfermedad hasta el momento.
No hay reportes publicados en nuestro medio sobre esta patología debido a la rareza de su presentación y a la dificultad para realizar estudios genéticos. Cuando un niño presenta falla renal aguda, anemia y trombocitopenia, sin pródromos de diarrea, y si además esto se presenta en forma recurrente, como es el caso de nuestro paciente, debe descartarse la posibilidad de SUH atípico, con la finalidad de iniciar un tratamiento rápido y oportuno. La guía elaborada por el Grupo de Estudio Pediátrico Europeo para SHU recomienda iniciar tratamiento con plasmaféresis tan pronto como sea posible, dentro de las 24 horas de presentación, en paralelo con tratamiento conservador (diálisis, transfusiones, antihipertensivos, etc.)(16). Esto, junto con la infusión de plasma fresco congelado, que reemplaza las proteínas defectuosas por proteínas funcionales, se considera la primera línea de tratamiento. Además, debido a que existe un desorden de sobreactivación del complemento, una opción terapéutica prometedora, tanto en la fase aguda como en la prevención de las recurrencias, es el uso de anticuerpos monoclonales humanizados dirigidos contra los componentes activadores claves de la ruta final del complemento como el C5 (eculizumab) (4,17). Estas medidas, pese a que aun existe debate con respecto a su eficacia, debido a que no hay estudios prospectivos controlados randomizados, han disminuido la tasa de mortalidad de 50 a 25% (3,6,17). Por lo tanto, un diagnóstico adecuado y tratamiento oportuno en estos casos puede cambiar el pronóstico de la enfermedad.
REFERENCIAS BIBLIOGRÁFICAS
1. Elliot E, Robins-Browne R. Hemolytic Uremic Syndrome. Curr Probl Pediatr Adolesc Health Care 2005; 35: 310-330. [ Links ]
2. Amirlak I, Amirlak B. Haemolytic uraemic syndrome: An overview. Nephrology 2006; 11:213-218. [ Links ]
3. Loirat C, Noris M, Fremeaux-Bacchi V. Complement and the atypical hemolytic uremic syndrome in children. Pediatr Nephrol 2008; 23: 1957-1972. [ Links ]
4.Hirt-Minkowski P, Dickenmann M, Schifferli J. Atypical Hemolytic Uremic Syndrome: Update on the Complement System and What is new. Nephron Clin Pract 2010; 114:219-235. [ Links ]
5.Johnson S, Taylor M. What´s new in haemolytic uraemic syndrome. Eur J Pediatr 2008; 167: 965-971. [ Links ]
6. Kavanagh D, Goodship T, Richards A. Atypical haemolytic uraemic syndrome. Br Med Bull 2006; 77-78:5-22. [ Links ]
7.Kavanagh D, Goodship T. Update in evaluating complement in hemolytic uremic syndrome. Curr Opin Nephrol Hypertens 2007; 16:565-571. [ Links ]
8.Kavanagh D, Richards A, Hauhart R, et al. Characterization of mutations in complement factor I associated with Hemolytic Uremic Syndrome. Mol Inmunol 2008; 45:95-105. [ Links ]
9.Bienaime F, Dragon-Durey MA, Regnier C, et al. Mutations in components of complement influence the outcome of Factor I-associated atypical hemolytic uremic syndrome. Kidney Internat 2010; 77:339-349. [ Links ]
10. Saunders RE, Abarrategui-Garrido C, Fremeaux-Bacchi V, et al. The interactive factor H-atypical hemolytic uremic syndrome mutation database and website: update and integration of membrane cofactor protein and Factor I mutations with structural models. Hum Mutat 2007; 28:222–234. [ Links ]
11. Carpioli J, Noris M, Brioschi S, et al. Genetcis of HUS: The impact of MCP, CFH and IF mutations on clinical presentation, response to treatment and outcome. Blood 2006; 108 (4):1267-1279. [ Links ]
12. Kavanagh D, Kemp E, Mayland E, et al. Mutations in complement factor I predispose to development of Atypical Hemolytic Uremic syndrome. J Am Soc Nephrol 2005; 16:2150-2155. [ Links ]
13. Sellier-Leclerc A, Fremeaux-Bacchi V, Dragon-Durey M, et al. Differential impact of complement mutations on clinical characteristics in atypical Hemolytic Uremic Syndrome. J Am Soc Nephrol 2007; 18:2392-2400. [ Links ]
14. Vyse TJ, Morley BJ, Bartók I, et al. The molecular basis of hereditary complement Factor I deficiency. J Clin Invest 1996; 97:925–933. [ Links ]
15. Zheng XL,Sadler JE. Pathogenesis of Thrombotic Microangiopathies. Annu Rev Pathol 2008; 3: 29-277. [ Links ]
16. Ariceta G, Besbas N, Jonson R, et al. Guideline for the investigation and inicial therapy of diarrea-negative hemolytic uremia síndrome. Pediatr Nephrol 2009; 24:687-696. [ Links ]
17. Waters AM, Licht C. aHUS caused by complement dysregulation: New therapies on the horizon. Pediatr Nephrol 2011; 26(1): 41-57. [ Links ]
Correspondencia:
Dr. Reyner Loza Munárriz
Unidad de Nefrología Pediátrica,
Hospital Nacional Cayetano Heredia
Av. Honorio Delgado s/n, San Martín de Porras, Lima, Peru
Teléfono: 3825906 Fax: 3825906
Correo electrónico: reyfe@hotmail.com
