










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Medica Herediana
Print version ISSN 1018-130X
Rev Med Hered vol.22 no.4 Lima Oct./dic. 2011
Conversatorio clínico patológico en el Hospital Nacional Arzobispo Loayza_2011-02.
Clinical Case at the Hospital Nacional Arzobispo Loayza_2011-2.
Editor Responsable: Dr. Enrique Cipriani1
Discusión Clínica: Dr. Roberto Aviles2
Discusión Patológica: Dr. Oscar Ruíz3
Residente Responsable: R3 Katherine Charaja Costa4, Leonardo Valladares4
1Profesor del Departamento de Medicina. Facultad de Medicina Alberto Hurtado Universidad Peruana Cayetano Heredia. Lima, Perú.
2Médico Asistente del Departamento de Medicina Hospital Nacional Arzobispo Loayza. Lima, Perú.
3Médico Asistente del Servicio de Medicina. Hospital Nacional Arzobispo Loayza. Lima, Perú.
4Médico Residente de Medicina. Universidad Nacional Mayor de San Marcos. Lima, Perú.
Enfermedad actual
Mujer de 25 años, natural de Lima, procedente de La Victoria, soltera, católica; Un mes y 10 días antes del ingreso inició su enfermedad caracterizada por dolor tipo cólico en el epigastrio irradiado a hipocondrio derecho, permanente, que disminuía en posición decúbito ventral, y parcialmente con analgésicos. Veinte días antes del ingreso notó tinte ictérico de piel y mucosas, y coluria. Una semana antes del ingreso presentó vómitos de contenido bilioso y el dolor se tornó insoportable.
Funciones biológicas: Apetito disminuido, sed aumentada, orina coluria, deposiciones normales, sueño aumentado y peso conservado.
Antecedentes Personales
Hospitalización por colecistitis aguda en 2008.
Colecistectomía en 2008.
G: 0 P: 0000
No alergia a medicamentos.
Examen Físico
PA: 110/80, FC: 72 x, FR: 18 x, T: 37°C
Buen estado general, palidez moderada, ictericia hasta abdomen, no tenía edemas. Tórax, pulmones y corazón normales. En abdomen había leve dolor a la palpación profunda en el epigastrio, y cicatriz de la colecistectomía; genitourinario normal. Al examen neurológico, estaba lúcida y orientada, no había signos meníngeos ni signos de focalización, tono muscular y los reflejos osteotendinosos eran normales.
Los resultados de los exámenes auxiliares al ingreso se muestran en la tabla 1. El sedimento urinario mostró: Leucocitos: 2 a 5 por campo, hematíes: 0 a 2 por campo, células epiteliales abundantes, y la radiografía de tórax era normal.

La ecografía abdominal mostró hígado de tamaño normal, bordes uniformes, parénquima homogéneo, vías biliares intrahepáticas no dilatadas, vena porta de 9 mm, y no había masas ni colecciones. Colédoco dilatado 20 mm, cálculos de 18 y 20 mm y vesícula ausente. Páncreas de volumen normal, aspecto heterogéneo por proceso inflamatorio. Conducto de Wirsung dilatado. Bazo aumentado de tamaño 20 cm, riñones normales. No había líquido libre en cavidad.
Discusión clínica:
Dr. Roberto Avilés
Es una mujer de 25 años, la edad indica que tiene una enfermedad congénita, hereditaria, metabólica o inmunológica, y voy a circunscribirme a ellas; no voy a pensar en neoplasias o enfermedades degenerativas ni infecciosas por los datos de la historia.
Dentro de los antecedentes personales, colecistitis aguda en el 2008, es un dato importantísimo. ¿Qué mujer tiene patología biliar a los 25 años? Aquí, la primera disyuntiva
con respecto a la utilidad de los antecedentes. ¿Será obesa?, ¿será gran multípara?, ¿usará anticonceptivos?, ¿ha tenido nutrición parenteral?, ¿ha usado hipoglicemiantes? Esos factores son los que coadyuvan a formar cálculos.
En este caso no existen estos antecedentes y es nulípara y no ha tenido gestaciones; pero no sabemos si ha usado métodos anticonceptivos. Tiene un IMC adecuado, por lo tanto la obesidad no es el factor en ella. Como no hay estos factores, hay que suponer que pueda tener una hemoglobinopatía o una alteración hemolítica crónica.
Los cálculos biliares más frecuentes en el mundo contienen colesterol como componente principal y los cálculos más raros son de pigmentos biliares. Los cálculos de colesterol son mucho más consistentes, múltiples, duros de contextura, a diferencia de los cálculos de pigmentos biliares que son frágiles.
El tiempo de enfermedad de un mes y medio, dolor tipo cólico en epigastrio con irradiación a hipocondrio derecho que cede en la posición de decúbito ventral, es una historia calcada de una pancreatitis aguda, es decir en la anamnesis, no hay dolor que se le parezca. Se debe descartar una isquemia mesentérica, una úlcera penetrante, o aneurisma de la aorta, pero como en medicina hay que tener en cuenta también la estadística, el dolor con esa característica más el antecedente biliar debe hacer pensar en pancreatitis de etiología biliar incluso antes de ver la ecografía. La ictericia de piel y mucosas, y la coluria, dan mayor asidero al diagnóstico.
En las funciones biológicas, el apetito disminuido puede tener varias connotaciones, tales como una enfermedad crónica; la patología biliar da hiporexia, la pancreatitis también la produce. La sed aumentada se explica por el cuadro emético, y no ingiere alimentos.
La información del examen físico es muy escueta y siempre digo que el examen físico es de la mayor utilidad. Si se encuentra ictericia, telangiectasias, eritema palmar, se enfoca a una hepatopatía crónica; si hay una vesícula palpable, se enfoca a una neoplasia y si se encuentra ictericia flavínica se piensa en una anemia hemolítica.
La dualidad anemia-ictericia es hemólisis hasta que se demuestre lo contrario. Esta asociación se puede observar en neoplasia de cabeza de páncreas, cáncer vesicular o cirrosis con discrasia sanguínea, hematemesis, o rectorragia, pero no hay evidencias de ellas en la historia de la paciente. Entonces, la historia primero nos habla de obstrucción y ahora, también de hemólisis.
¿Por qué este paciente ha vuelto a hacer cuadro obstructivo, si le realizaron colecistectomía? Hay dos fenómenos, la litiasis residual y la litiasis recurrente. La litiasis residual ocurre dentro de los dos años de operado. Pero viendo la cronología de la enfermedad de la paciente, hay un intervalo de más de dos años. Si fuera litiasis residual la primera causa sería la migración de un cálculo de la vesícula a las vías biliares, formando el síndrome del bacinete (Mirizzi). Si fuera litiasis recurrente tendría más asidero el diagnostico que plantearé al final; porque la litiasis recurrente, implica la formación de nuevos cálculos, que se pueden formar en el colédoco, pero que por el mecanismo fisiopatológico de presión del colédoco que existe a nivel de la ampolla de Vater y de los conductos, hace que estos cálculos sean de otras características.
¿Qué enfermedades podrían condicionar la litiasis recurrente? Hay una enfermedad que se presenta en gente joven, la enfermedad de Caroli, que es una colestasis intrahepática crónica; la cirrosis biliar primaria que es una enfermedad inmunológica y también se debe considerar la colangitis esclerosante.
En los exámenes auxiliares la hemoglobina era 9,1 gr/dL, que se considera anemia leve a moderada; se puede ver en pancreatitis aguda, ictericia obstructiva, litiasis residual, anemia por enfermedad crónica, anemia ferropénica probablemente. ¿Por qué anemia ferropénica?, como es una mujer joven, no sé si hay hipermenorrea o menometrorragia, pero la primera causa de anemia en mujeres es ferropénica, y de esta la primera causa es ginecológica.
El hemograma con 6 400 leucocitos y plaquetas normales, aleja la posibilidad de anemias aplásicas, hipoplásicas o mieloptisis y las anemias congénitas de Fanconi en las hay repercusión en el paciente sobre todo en las plaquetas, coagulación, incluso en los leucocitos. No hay bicitopenia o pancitopenia para pensar en anemia megaloblástica.
El tiempo de protrombina en 16,5 seg es un hallazgo importante; aquí dos disyuntivas son: ¿por hepatopatía? ¿por alteración en la síntesis de factor de coagulación? o ¿es por la obstrucción? Pienso que debe haber algo de ambos.
La administración parenteral de vitamina K por varios días, corrigen el tiempo de protrombina. La urea, creatinina, glucosa y proteínas en sangre eran normales; nos abocaremos a la interpretación de la hiperbilirrubinemia. El enfoque se debe hacer asociándola con los niveles de transaminasas y fosfatasa alcalina; la paciente tiene una hiperbilirrubinemia mixta, 4,34 mg/dL de bilirrubina directa y 2,4 mg/dL de bilirrubina indirecta, que se puede ver en hepatopatías, problemas crónicos obstructivos, hemólisis crónicas, y una serie de patologías que pueden dar hiperbilirrubinemia directa.
En relación con las transaminasas, la TGO es menos específica para hepatopatía, pero más específica para otras causas. La TGP es más específica de hepatopatía; en la paciente vemos que el índice TGO/TGP está elevado; se podría decir que la TGO está elevada pues ello ocurre en la pancreatitis aguda y en la hemólisis. El aumento de amilasa es un buen índice, es muy sensible pero no tan específica como la lipasa. La paciente tiene una amilasa más de 5 a 6 veces lo normal, esto es consistente con una pancreatitis aguda de etiología biliar; y la lipasa positiva lo confirma.
Lo que llama la atención en este caso es que la fosfatasa alcalina está elevada solo dos y medio veces su valor normal; en mujeres debería ser mayor debido a que hay un cuadro obstructivo, pero algunas veces no hay obstrucción total sino parcial, es por eso que la fosfatasa alcalina no se eleva tanto. No hay correlación entre el grado de obstrucción con los niveles de fosfatasa alcalina porque puede haber otras alteraciones extrahepáticas que pueden elevarla, por eso es importante medir la gamma glutamil transpeptidasa.
La ecografía abdominal reveló dilatación del colédoco que correlaciona con su cuadro clínico. Llama la atención el colédoco en 20 mm, esto es lo que se denomina megacolédoco. El megacolédoco se ve cuando hay cálculos mayores de 5 mm o por obstrucción por neoplasia y algunos parásitos; esto es importante y de utilidad para el gastroenterólogo o cirujano que realizará la colangiografía endoscópica retrograda o la cirugía. Además, se encontró esplenomegalia.
La esplenomegalia gigante tiene tres causas: metaplasia mieloide agnogénica, linfoma y enfermedad infiltrativa (1).
En orden descendente mi primer diagnóstico sería una anemia hemolítica hereditaria; cuyo diagnóstico diferencial incluye:
a. Defectos de membrana;
b. Defectos enzimáticos;
c. Hemoglobinopatías y;
d. Hemoglobinuria paroxística nocturna.
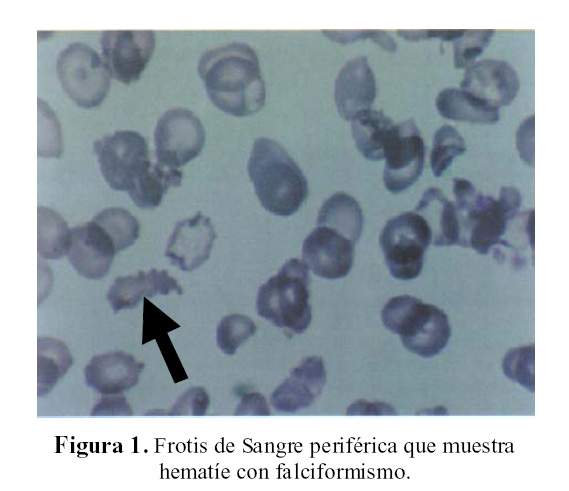
¿Será una anemia falciforme? (2). Una característica de las anemias falciformes es el dolor óseo, dolor cefálico, dolor en las extremidades, dolor esplénico, cuadro venoclusivo con crisis, e infarto esplénico. Por la ausencia de falciformismo en el frotis de sangre periférica (Figura 1) y por la ausencia de dolor, esta posibilidad se aleja.
Las talasemias se caracterizan por microcitosis; la a y b talasemias son raras. No se debe dejar de pensar en las anemias hemolíticas autoinmunes, que pueden ser primarias o secundarias (3). Las primarias tienen test de Coombs positivo, con hemoglobina disminuida, DHL y reticulocitos aumentados, pero no hay gran esplenomegalia; las secundarias son de causa infecciosa, linfoproliferativa, o colagenopatías (2).
Mi posibilidad diagnóstica definitiva es esferocitosis hereditaria. Existen presentaciones típicas, leves o severas además de los portadores asintomáticos; pienso que la paciente es portador asintomático puesto que se presentó en la edad adulta.
Discusión Patológica
Dr. Oscar Ruiz Franco
La madre de la paciente tiene anemia hemolítica no tipificada. Se ha mencionado que la paciente acudió a este hospital por un cuadro de litiasis vesicular, que fue operada. Dos años después ingresó por pancreatitis y nuevamente litiasis pero esta vez en el colédoco; el CEPRE evidenció cálculos que no se pudieron extraer, siendo reintervenida quirúrgicamente extirpándosele dos cálculos del colédoco.
El volumen corpuscular medio por encima de 35 hace pensar en una anemia hemolítica tipo esferocitosis hereditaria (Figura 2); entonces desde el momento que se tienen las constantes corpusculares puede hacerse una diferenciación.
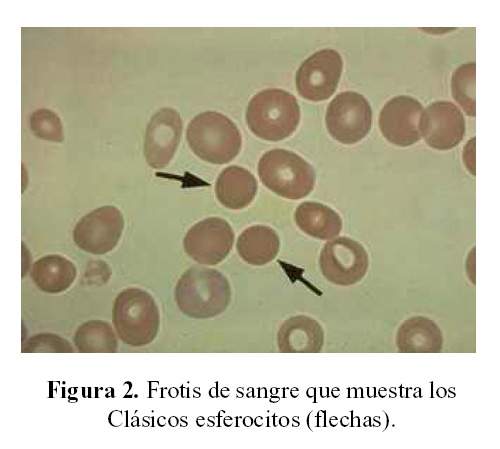
En el frotis de sangre periférica se encontró anisopoiquilocitosis, un hallazgo totalmente inespecífico, pero llamaba la atención la presencia de microesferocitos, que son glóbulos rojos, redondos, muy pequeños. Cuando se encuentran por encima del 10%, la primera posibilidad es esferocitosis hereditaria. Pueden estar presentes en anemia hemolítica de tipo autoinmune, también en la talasemia, sobre todo en los portadores; además, ayudaba la presencia de policromatofilia y de punteado basófilo, que son signos de regeneración medular, que se ven en los cuadros hemolíticos. Había polimorfonucleares polisegmentados (más de 5 lóbulos), que indica la existencia de un componente megaloblástico puesto que hay exceso de consumo de folato.
El test de Coombs directo fue negativo, lo que de plano dice que la anemia hemolítica no es de tipo autoimune.
Un test de fragilidad globular, hecho hace siete años estaba dentro de rango normal. ¿En qué consiste este test de fragilidad globular? Consiste en exponer sangre con anticoagulante a diferentes concentraciones de cloruro de sodio para ver a que concentración el glóbulo rojo se destruye o hay expresión de cuadro hemolítico. Normalmente en las personas que no tienen alteración a nivel de la membrana del glóbulo rojo la hemólisis se produce entre 0,45 y 0,35. Y de hecho el test de fragilidad osmótica fue negativo, por lo tanto puede negar que el paciente tenga esferocitosis hereditaria, porque este es el "gold standard" para esferocitosis hereditaria; aunque no siempre es positivo (4). Lo que no sé, es si este valor corresponde al que se hizo a las 24 horas de haber sido extraída la muestra, porque el test de fragilidad osmótica, de fragilidad globular se tiene que hacer en dos tiempos: a la hora de haber sido extraída la muestra, y la prueba confirmatoria que se hace luego de haber incubado la sangre a 37°C por un día. Podemos suponer que al dar ese resultado sería de la muestra incubada, porque usualmente se dan los dos valores.
Esferocito es simplemente un glóbulo redondo, microesferocitos son glóbulos rojos redondo pero muy pequeños, que es más característico de la esferocitosis hereditaria, y para que estos se presenten debe existir un acondicionamiento en el bazo que lo hace más pequeño, como veremos después.
Los reticulocitos estaban altos y el hematocrito estaba por debajo de lo normal. En la ecografía abdominal y también me parece por el hallazgo intraoperatorio encontraron esplenomegalia; entonces, anemia, ictericia crónica ligadas a la presencia de esplenomegalia va de la mano con anemia hemolítica, y como se ha presentado en una persona joven, lo más probable es que se trate de una anemia hemolítica congénita.
El diagnóstico diferencial sería, en primer lugar por los hallazgos del frotis de la sangre periférica, esferocitosis hereditaria: En primer lugar, mujer joven con anemia asociada a ictericia, reticulocitosis que confirma un cuadro hemolítico. En segundo lugar, la presencia de litiasis vesicular en una joven, sin antecedentes de multiparidad. ¿Por qué ocurre litiasis vesicular en pacientes con hemólisis crónica? ¿Por qué es posible que se encuentren estos hallazgos en niños de 4 a 5 años? Porque, hay microtrombosis a nivel de la vesícula biliar y de los conductos biliares; hay lentitud del flujo de la bilis en los conductos biliares y; hay disminución de la cantidad de sales.
Otro punto y más interesante, es la presencia de microesferocitos por encima de 10%, además la concentración de la hemoglobina corpuscular media que se mantiene en promedio por encima de 35.
En el hematíe, la principal función de la membrana celular es mantener la forma, dada por su flexibilidad que se la da la integridad de las proteínas de la membrana; de tal manera que el glóbulo rojo normal se puede adaptar a cualquier calibre de los capilares.
La prueba confirmatoria como hemos dicho es el test de la fragilidad osmótica que se debe hacer en dos momentos: a la hora y a las 24 horas; hubiese sido muy interesante hacerle en nuestro hospital el test de fragilidad osmótica.
Para el diagnóstico de la anemia, la hemoglobina es más estable en su determinación. No sufre cambios como si puede ocurrir con el hematocrito, como por ejemplo puede estar falsamente elevado en la deshidratación o en un paciente con hipocromía. En la anemia microcítica/hipocrómica se pierde la relación 3:1 entre hematocrito y la hemoglobina.
En los casos de pérdida de la relación 3:1 entre hematocrito y hemoglobina, hay que evaluar la hemoglobina corpuscular media. Si es menor de 27 hay hipocromía, por lo tanto es cierto que hay una pérdida de esta relación. La causa más frecuente de pérdida normal de la relación 3:1 entre hematocrito y hemoglobina en Perú y en el mundo es la anemia ferropénica.
La anemia más frecuente en el mundo es definitivamente la ferropénica porque está ligada al desarrollo socioeconómico y a los nutrientes que dan hierro. La hemoglobina es una proteína compleja que tiene dos elementos importantes, el núcleo que tiene una cadena proteica llamada protoporfirina y el hierro en estado reducido (valencia 2), para captar el oxígeno, y una fracción netamente proteica, la globina. La globina tiene determinación genética, las dos cadenas alfas en el cromosoma 16 y las dos cadenas no alfa en el cromosoma 11. Las cadenas no alfa dan el nombre a los tipos de hemoglobinas normales que tenemos.
Las hemoglobinas normales son: la hemoglobina A1 con dos cadenas alfa y dos cadenas beta, que constituye hasta el 98%; la hemoglobina fetal constituida por dos cadenas alfa y dos cadenas gama, cumple una función importante durante la vida intrauterina, aunque también la podemos tener de adultos, y la hemoglobina A2, que tiene dos cadenas alfa y dos cadenas delta. La hemoglobina fetal puede representar hasta 2% y la hemoglobina A2 hasta 3,5%. Si la hemoglobina A2 es mayor de 3,5% estaríamos frente a una talasemia, siendo un marcador de talasemia sobre todo en la b-talasemia.
La anemia hemolítica que es el motivo de la discusión, se puede dividir en dos grupos: congénitas o intracorpusculares en las que el defecto está en el glóbulo rojo, pudiendo a nivel de la membrana como la esferocitosis hereditaria; estar a nivel enzimático como en la deficiencia de glucosa-6-fosfato deshidrogenasa o de piruvato quinasa; o las hemoglobinopatías, y adquiridas, sobre todo las de tipo inmune.
La clínica de la hemólisis se caracteriza por la triada: anemia, ictericia y hepato-esplenomegalia, sobretodo esplenomegalia porque es el bazo el órgano hemocaterético por excelencia, donde se van a destruir todas las células sanguíneas que tienen alguna alteración. ¿Cuándo se sospecha un cuadro hemolítico crónico o congénito? Cuando existe litiasis vesicular en jóvenes, que es totalmente inusual. Pero también, cuando haya ulceras en las partes distales de las extremidades, entonces estaríamos frente a la anemia drepanocítica o enfermedad homocigota de la hemoglobina S. Las alteraciones esqueléticas se pueden presentar en todas las formas de anemia hemolítica congénita dependiendo de la severidad del cuadro, y el retardo en el crecimiento, lo mismo. Pero, la patología que más afecta el crecimiento es la talasemia.
En el laboratorio podemos sospechar anemia hemolítico, cuando hay policromatofilia, punteado basófilo, presencia de glóbulos rojos nucleados o normoblastos, disminución de la haptoglobina, incremento de la deshidrogenasa láctica, hiperbilirrubinemia a predominio indirecto y en algunos casos hay hemoglobinuria. Hemoglobinuria ocurre cuando la hemolisis ocurre dentro del vaso sanguíneo, como por ejemplo en la hemoglobinuria paroxística nocturna.
Entonces por los hallazgos estaríamos frente a una anemia hemolítica congénita tipo membranopatía (CHCM mayor de 35, presencia de microesferocitos asociado a policromatofilia y punteado basófilo).
En la esferocitosis hereditaria la causa es la anormalidad en una o más proteínas de la membrana del glóbulo rojo que sirven de anclaje entre las dos capas lipídicas; como consecuencia hay liberación de lípidos, el glóbulo rojo se vuelve más pequeño, pierde la forma bicóncava y se hace esférico.
Entre las causas de esferocitosis hereditaria (5) está la deficiencia parcial de espectrina, que es una de las proteínas que sirven de anclaje; la deficiencia de anquirina, aunque es más frecuente la deficiencia combinada de espectrina y anquirina; y las proteínas de la banda 3 y las proteínas 4.2.
Luego de formados los microesferocitos, los hematíes llegan al bazo donde se va a expresar en mayor intensidad el defecto y dejan pasar a los más pequeños que no son destruidos, por eso se conoce con el nombre de microesferocitos, frecuentes en la esferocitosis hereditaria.
En el frotis de sangre periférica de una persona con esferocitosis hereditaria vamos a encontrar: glóbulos rojos redondos, pequeños y no tienen la palidez central característica que no debe exceder a la tercera parte de toda la superficie. Podemos encontrar glóbulos rojos mellados o en pinza que son característicos cuando hay deficiencia de proteína de la banda 3, acantocerocítos cuando falta la proteína b-espectrina y contorno irregular cuando falta la espectrina y anquirina.
Otras pruebas que pueden ayudar al diagnóstico de esferocitosis hereditaria son, el test de autohemolisis y unas pruebas muy especializadas en las que hay necesidad de contar con un ectacitómetro que mide la rigidez del glóbulo rojo, y por último el análisis de las proteínas para saber cuál es la que está involucrada en la génesis de la esferocitosis, pruebas que en Perú no las tenemos aún, pero con la fragilidad osmótica es más que suficiente, hasta ahora esa sigue siendo la prueba confirmatoria del diagnóstico.
REFERENCIAS BIBLIOGRÁFICAS
1. Henry P, Longo D. Enlargement of Lymph nodes and spleen. En: Harrisons Internal Medicine. Mc Graw Hill. 16th. Edition. 2005. p.343-348. [ Links ]
2. Ideguchi H, Ishikawa A, Futata Y, et al. A comprehensive scheme for the systematic investigation of hemolytic anemia. Ann Clin Lab Sci 1994; 24(5): 412-21. [ Links ]
3. Hashimoto C. Autoimmune hemolytic anemia. Clin Rev Allergy Immunol 1998; 16(3):285-95. [ Links ]
4. Korones D, Pearson HA. Normal erythrocyte osmotic fragility in hereditary spherocytosis. J Pediatr 1989; 114(2):264-6. [ Links ]
5. Agre P, Asimos A, Casella JF, McMillan C. Inheritance pattern and clinical response to splenectomyerythrocyte spectrin deficiency in hereditary spherocytosis. N Engl J Med 1986; 315(25):157. [ Links ]
