










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Medica Herediana
versión impresa ISSN 1018-130X
Rev Med Hered v.23 n.1 Lima mar. 2012
Gran Ronda de Medicina Interna y especialidades del Hospital Nacional Cayetano Heredia.
Caso clínico 01-2012. Varón de 81 años de edad con disnea progresiva, edema de miembros inferiores y orina espumosa.
Grand Round of Internal Medicine and specialties at the Hospital Nacional Cayetano Heredia. Clinical case 01-2012. Male 81 years old with progressive dyspnea, leg edema and foamy urine.
Editor de sección: Dr. Sergio Vásquez Kunze; Editores asociados: Dr. Héctor Sosa Valle, Dr. Ray Ticse Aguirre, Dra. Elena Zelaya Arteaga
Delia Alva 1, Héctor Sosa2, Sergio Vásquez2, Martín Salazar3, Oscar Gayoso 4, Cristian León 5, Miro Rodríguez 6
1Médico Residente de 3° año de Medicina Interna. Dpto. de Medicina, Hospital Nacional Cayetano Heredia. Lima, Perú.
2Médico asistente, Servicio de Medicina Interna. Dpto. de Medicina, Hospital Nacional Cayetano Heredia. Lima, Perú.
3Jefe del Servicio de Cardiología. Dpto. de Medicina, Hospital Nacional Cayetano Heredia. Lima, Perú.
4Médico asistente, Servicio de Neumología. Dpto. de Medicina, Hospital Nacional Cayetano Heredia. Lima, Perú.
5Médico asistente, Servicio de Nefrología. Dpto. de Medicina, Hospital Nacional Cayetano Heredia. Lima, Perú.
6Médico asistente, Servicio de Hemato-Oncología. Dpto. de Medicina, Hospital Nacional Cayetano Heredia. Lima, Perú.
PALABRAS CLAVE: Amiloidosis, mieloma múltiple, síndrome nefrótico (Fuente: DeCS BIREME).
KEYWORDS: Amyloidosis, multiple mieloma, nephrotic syndrome. (Fuent: MeSH NLM)
CASO CLÍNICO
Dra. Delia Alva Rodríguez (residente de tercer año de medicina interna)
Varón de 81 años natural y procedente de Huancayo que ingresó a nuestro hospital con historia de 2 meses de evolución caracterizada por disnea progresiva, aumento de volumen en miembros inferiores y orina espumosa.
Dos semanas antes del ingreso la disnea progresó de grandes a moderados esfuerzos y se agregaron roncantes, sibilancias y edema de miembros inferiores. No había presentado disnea paroxística nocturna, palpitaciones, dolor precordial, síncope o disminución de volumen urinario. Por estas molestias acudió al consultorio externo de neumología. El examen físico en esa consulta mostró una presión arterial en 100/60 mm Hg, frecuencia respiratoria en 24 por minuto, frecuencia cardiaca en 90 por minuto, estaba afebril, se encontró subcrépitos en bases bilateral y sibilancias en el examen de pulmones, había ingurgitación yugular y edema que dejaba fóvea en miembros inferiores. El resto del examen no fue contributorio.
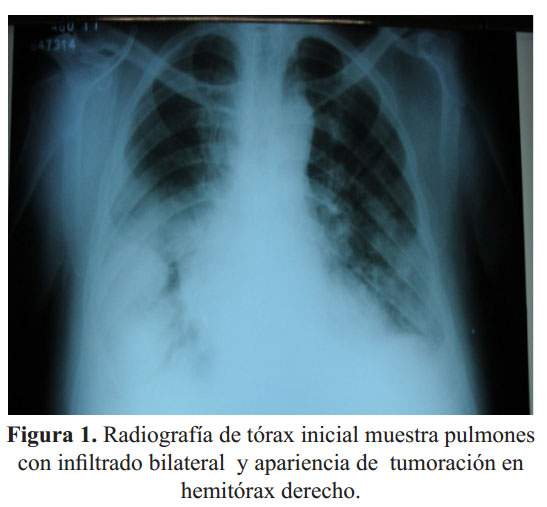
Se le realizó una radiografía de tórax ( Figura 1) y tomografía de tórax, esta última mostraba engrosamiento de septos interlobulillares, efusión loculada en cisura horizontal y oblicua del pulmón derecho, infiltrado intersticial a predominio de bases y efusión pleural bilateral. Por hallazgos de enfermedad obstructiva se prescribieron corticoides, ß2-agonistas y se solicitó NT-proBNP por sospecha de insuficiencia cardiaca.
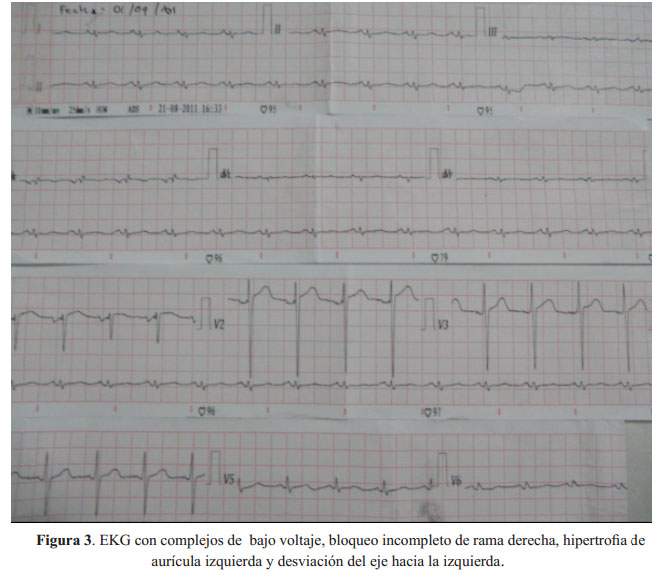
Fue evaluado en el consultorio de cardiología donde se le realizó un electrocardiograma que mostró complejos de bajo voltaje, bloqueo incompleto de rama derecha, hipertrofia de aurícula izquierda y desviación del eje hacia la izquierda. El ecocardiograma, mostró una fracción de eyección (FE) de 54%, hipertrofia ventricular izquierda moderada concéntrica, esclerosis valvular mitral y aórtica de grado moderado con apertura valvular normal. El estudio Doppler mostró dilatación leve de la aurícula izquierda, flujo mitral con patrón de disfunción diastólica seudonormalizado y regurgitación moderada a severa, insuficiencia aórtica moderada e hipertensión pulmonar leve. El resultado del NT-pro BNP fue 4,690 pg/mL.
Por la historia de orinas espumosas fue evaluado en el consultorio de nefrología. Se encontró proteinuria en 24 horas de 3552 mg/día y depuración de creatinina en 44 ml/min, el examen de orina mostró densidad en 1010, pH 5,0, cilindros granulosos 1+ y no había hematuria ni leucocituria; el urocultivo fue negativo. El proteinograma electroforético de orina fue: albúmina 3,01 gr, α-globulinas 0,01 gr β-globulinas 0,17 y gamma-globulinas 0,22 gr. Ademαs, VSG 49 mm/h, C3: 97 mg/dL (VN: 86-184), PSA 2,40 mcg/L (VN < 4), ANA, ANCA, serología para brucela y proteinuria de Bence Jones negativos y perfil lipídico, TSH y T4 libre normales. La ecografía renal mostró riñones de 8,9 cm y 8,8 cm de longitud, con parénquima de ecogenicidad conservada y próstata postquirúrgica sin alteraciones. El paciente recibió furosemida y fue controlado ambulatoriamente.
Una semana antes del ingreso el edema en miembros inferiores se incrementó, tres días antes del ingreso la disnea empeoró a leves esfuerzos y hasta no tolerar el decúbito, por lo que fue llevado a emergencia de nuestro hospital, siendo admitido.
El apetito estaba disminuido, el sueño alterado por la tos y la disnea, la diuresis era de aproximadamente 1,5 L al día y "espumosa", las deposiciones eran normales y había disminuido un kilogramo en dos meses.
No había historia de hipertensión arterial, diabetes, tuberculosis y contactos tuberculosos. Prostatectomía hacía 18 años, por hiperplasia benigna de próstata. Negaba reacciones medicamentosas, uso de fármacos, viajes, etilismo, tabaquismo y antecedentes familiares de importancia. Era un paciente completamente funcional.
El examen clínico al ingreso mostró PA 100/60mmHg, frecuencia cardíaca 103 x, frecuencia respiratoria 23 x, SO2: 93%, T: 37°C y peso 67 Kg. Estaba hidratado, despierto y orientado en las tres esferas y no presentaba fascies característica, La piel era tibia, elástica y no había palidez ni ictericia. Había edema de miembros inferiores 2+/3+. No había linfadenopatias. En pulmones, el murmullo vesicular estaba disminuido en ambos campos pulmonares con crepitantes y subcrepitantes difusos; los ruidos cardiacos eran rítmicos y taquicárdicos, de buena intensidad, no había soplos, pero tenía ingurgitación yugular. El abdomen era blando y no se palpaba visceromegalia y al examen neurológico no había signos de focalización.
La radiografía de tórax mostraba infiltrados en ambos campos con redistribución de flujo de la vasculatura pulmonar. Los exámenes de laboratorio al ingreso y en los días posteriores, se muestran en la
tabla 1.

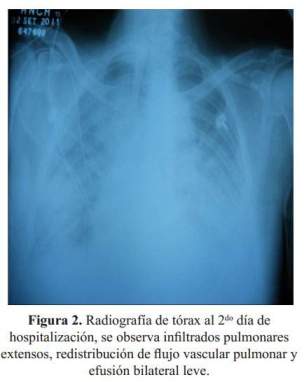
El paciente ingresó al servicio de medicina indicándosele furosemida, ceftriaxona, claritromicina y restricción de fluidos. A las pocas horas presentó súbitamente exacerbación de la disnea, requiriendo oxígeno por máscara de reservorio. Se le realizó una radiografía de tórax (
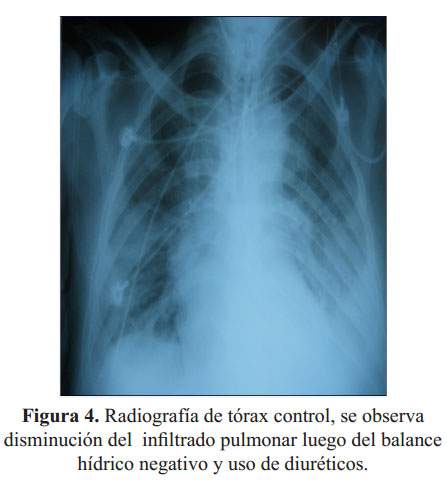
Figura 2), EKG ( Figura 3) y análisis de gases arteriales y fue transferido a la unidad de cuidados intensivos (UCI); se agregó enoxaparina ante la sospecha de tromboembolia e infarto de miocardio sin elevación del segmento ST. El paciente mejoró desde el punto de vista respiratorio clínica y radiográficamente ( Figura 4). Luego de cinco días salió de alta de la UCI, en el servicio de medicina interna se le realizó ecocardiografía la que mostró una FE de 48%, sin dilatación de cavidades, función sistólica del ventrículo izquierdo conservado con patrón restrictivo del flujo mitral.
DISCUSIÓN
La discusión se circunscribió en el diagnóstico etiológico y sobre otros procedimientos o exámenes auxiliares a realizar para llegar al diagnóstico.
Dr. Héctor Sosa Valle (Medicina Interna)
Teniendo en consideración el cuadro clínico, exámenes y antecedentes, el paciente presenta síndrome nefrótico e insuficiencia cardiaca. En adultos mayores con síndrome nefrótico se debe considerar la posibilidad de síndrome paraneoplásico o amiloidosis. La segunda opción es más plausible en este paciente porque además presenta insuficiencia cardiaca congestiva debido a una cardiopatía restrictiva; sin embargo, no hay evidencia de neuropatía que es bastante común en amiloidosis, pero no la excluye. De ser amiloidosis, lo más probable sería de tipo AL por tener el paciente más de 80 años y tener disfunción de más de un órgano (falla cardiaca y síndrome nefrótico) (1).
Para el diagnóstico requeriría estudios de inmunofijación sérica y urinaria buscando gammapatía monoclonal que se asocia a amiloidosis AL. El diagnóstico definitivo se establecería con biopsia de tejido de mucosa labial o de grasa periumbilical (esta última tiene mayor rendimiento diagnóstico) con tinción con colorantes supravitales como Rojo de Congo y con inmunohistoquímica de los depósitos de amiloide encontrados en los tejidos (2).
De considerarse la opción de síndrome paraneoplásico se debería ampliar el estudio con examen de aspirado de medula ósea (también sería útil en la evaluación de amiloidosis), endoscopía, colonoscopia y en último caso biopsia renal. Hay que mencionar que las biopsias de estos órganos también ayudarían al diagnóstico de amiloidosis.
Dr. Martín Salazar Cáceres (Cardiología)
El paciente fue evaluado a solicitud de neumología. Este refería disnea y edema de un mes de evolución, sin ortopnea ni disnea paroxística nocturna, que mejoró inicialmente con el uso de broncodilatadores. El EKG inicial mostró ritmo sinusal, complejos de bajo voltaje, bloqueo incompleto de rama derecha, hipertrofia de aurícula izquierda y desviación del eje hacia la izquierda, y el ecocardiograma, hipertrofia ventricular, función sistólica conservada, dilatación auricular y un patrón de flujo mitral de tipo seudonormalizado (restrictivo).
La presencia de complejos pequeños en el EKG y paredes engrosadas en la ecocardiografía sugiere fuertemente una cardiomiopatía de tipo restrictivo, siendo la más frecuente, amiloidosis. El hallazgo más característico de amiloidosis en el ecocardiograma, es un patrón moteado en la textura del miocardio, hallazgo que no estaba presente en el paciente.
En el tratamiento de amiloidosis, se debe utilizar diuréticos y restricción hídrica. Los pacientes con eta patología tienen muy pobre tolerancia al uso de Inhibidores de la enzima convertidora de angiotensina (IECA), beta-bloqueadores y digital, para el manejo de la insuficiencia cardiaca.
En amiloidosis sistémica con compromiso cardiaco la presencia de NT-proBNP elevado y en especial la elevación de troponina, le confiere mal pronóstico a corto plazo (3,4).
Dr. Oscar Gayoso Cervantes (Neumología)
Conocí el caso en el consultorio de neumología donde ya refería disnea y sibilancias siendo manejado como EPOC y que por posterior evidencia de insuficiencia cardiaca (IC) fue referido al servicio de cardiología.
Ante la presencia en la radiografía de tórax de una aparente masa pulmonar ( Figura 1), se solicitó una TEM de tórax evidenciándose la masa correspondía a una "tumoración fantasma" por IC. Aquí cabe mencionar la importancia del NT-proBNP en el diagnóstico de IC vs enfermedad pulmonar. En este paciente el NT-proBNP muy elevado indicaba que el cuadro tenía origen cardiaco. También se encontró líquido pleural, sin embargo, debido a la escasa cantidad, la toracocentesis fue diferida.
Las radiografías al ingreso al hospital muestran infiltrado bilateral y redistribución de flujo pulmonar sin cardiomegalia, lo que sugiere un edema agudo de pulmón cardiogénico por IC diastólica. El cuadro de IC diastólica, un EKG de complejos pequeños y proteinuria masiva, sugieren la posibilidad de una enfermedad sistémica con compromiso cardiaco y renal, como sería el caso de una amiloidosis. Otra posibilidad es fibroelastosis subendocardica de Loeffler, pero al no haber presencia de eosinofilia se aleja este diagnóstico. También se plantearon otras etiologías como linfoma, sarcoidosis o histiocitosis, sugiriéndose biopsia de mucosa, como parte del apoyo al diagnóstico para amilioidosis y proteinograma electroforético.
Dr. Cristian León Rabanal (Nefrología)
Estamos frente a un paciente con enfermedad renal crónica asociado a proteinuria masiva. Es tentador hablar de síndrome nefrótico; sin embargo, resaltar ese diagnóstico nos pone en riesgo de dejar de lado la importancia del deterioro de la función renal como el eje fundamental del diagnóstico y la proteinuria como un marcador de severidad y progresión de enfermedad renal crónica.
En el grupo etario del paciente, el síndrome nefrótico en causado en la mayoría de casos por enfermedad glomerular secundaria y por enfermedades neoplásicas que cursan con proteinuria como una manifestación paraneoplásica.
En este contexto, linfoma puede manifestarse como síndrome nefrótico con nefrosis lipoidea (cambios mínimos). Esta se presenta con proteinuria selectiva donde se produce filtración de proteínas como la albúmina, pero no las mas grandes como inmunoglobulinas (proteinuria selectiva: albúmina es mayor del 80%), que coincide con los hallazgos del proteinograma en orina del paciente (89% era albumina).
La otra posibilidad dado el compromiso multisistémico (corazón, riñón) es la amiloidosis sistémica; pero esta patología se manifiesta con proteinuria no selectiva (proporción de albúmina en el proteinograma electroforético menor al 80%) (5-7), característica de los procesos que cursan con daño en la estructura histológica del glomérulo (glomerulopatías severas en cuya orina la proporción de proteínas refleja la proporción plasmática). La proteinuria no selectiva se suele asociar a mayor riesgo de deterioro de la función renal.
Considerando que el paciente impresiona tener proteinuria selectiva se debe buscar patología neoplásica (como linfoma). De plantearse amiloidosis dado el compromiso multisistémico, se debe hacer los estudios específicos mencionados por los que opinaron previamente, antes de proceder a la biopsia renal.
Dr. Amador Carcelén Bustamante (Medicina Interna)
Esta presentación tiene un solo diagnóstico: Amiloidosis sistémica. La proteinuria en rango nefrótico y la insuficiencia cardiaca es característica. Llama la atención la ausencia de neuropatía autonómica; sin embargo, a veces puede estar ausente. El pronóstico es malo a pesar de la quimioterapia u otros esfuerzos terapéuticos. El diagnóstico puede tener dificultad, pues en la amiloidosis primaria los colorantes supravitales pueden no teñir el amiloide, y se podrían necesitar varias biopsias, de grasa periumbilical, rectal o biopsia de hueso. Se debe hacer también inmunofijación para hallar el pico monoclonal ya que el proteinograma electroforético tiene baja sensibilidad para su detección.
CONCLUSION
Se concluyó que el diagnóstico más probable era amiloidosis sistémica AL, debiéndose realizar tinciones para amiloide en la biopsia de labio o de grasa periumbilical y de no ser positivas, de medula ósea y de mucosa rectal. La biopsia de riñón quedaría como última alternativa dada la propensión al sangrado de los pacientes con amiloidosis. Además, se debe realizar inmunofijacion en sangre y orina para la búsqueda de cadenas ligeras monoclonales que apoyarían un diagnóstico de amiloidosis AL. Nefrología planteó también el diagnóstico de neoplasia oculta con síndrome nefrótico como manifestación paraneoplásica dada la selectividad de la proteinuria que es característica de cambios mínimos y su asociación con linfoma, a esta edad.
Evolución
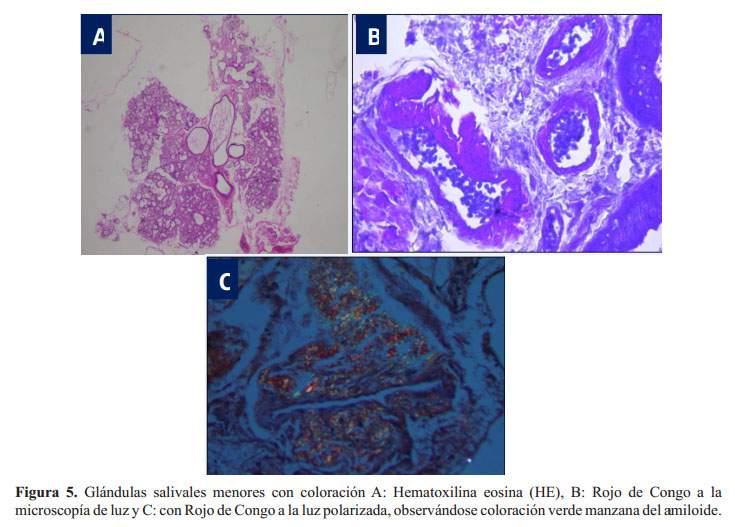
Se tomaron muestras de glándula salival menor que fueron revisadas por el Dr. Wilson Delgado A. (Facultad de Estomatología de la Universidad Peruana Cayetano Heredia) quien encontró algunos conductos salivares con engrosamiento de sus paredes debido a un material eosinófilo acelular con coloración para amiloide positivas (Rojo Congo a la luz polarizada) ( Figura 5), haciendo el diagnóstico de amiloidosis de la glándula salival menor. Posteriormente, en el estudio de inmunoelectroforesis por inmunofijación en sangre se detectó gamapatía monoclonal IgA lambda.
Fue dado de alta con furosemida, captopril, atorvastatina, bromuro de ipatropio y referido al consultorio de hemato-oncología para completar los estudios con aspirado de médula ósea (AMO) y proceder a quimioterapia, la cual el paciente decidió seguir en otro centro.
COMENTARIOS
Dr. Sergio Vásquez Kunze (Medicina Interna)
Existían 2 problemas pendientes a discutir en este caso. El primero, si realmente era una amiloidosis AL, dado que la prevalencia de encontrar un pico monoclonal a esta edad (gamapatia monoclonal de significado incierto) es de 3% a 8,9% (8), y que simplemente esa cadena ligera pueda no ser la causa de la amiloidosis de este paciente como se observa en muchos otros reportes (9).
Se sabe que el amiloide se forma por la configuración beta hoja plegada que pueden adoptar diversas proteínas, como la proteína A, la transtiretina o cadenas ligeras de inmunoglobulina. Se tiene que demostrar cuál de estas proteínas forman el tejido amiloide. Esto se puede hacer con paneles de anticuerpos con la técnica inmunogold en microscopía electrónica o un análisis de espectroscopia de masa (prueba de oro) en la biopsia (10).
En la actualidad, estas pruebas son claves en el diagnóstico definitivo del tipo de amiloide. Se hacen 4 tinciones: cadenas ligeras lambda, kappa, proteína A y transtiretina (11). Si la tinción fuera positiva para IgA lambda entonces es diagnóstica (Amiloidosis AL). Sin embargo, este paciente no tiene causas para amiloidosis secundaria (AA) y ésta casi nunca compromete el corazón. La amiloidosis por transtiretina no puede rechazarse del todo. Los dos tipos de transtiretina que producen amiloidosis son, la amiloidosis senil cardiaca (transtiretina salvaje) y la familiar de inicio tardío (transtiretina mutante) (2,12). La senil cardiaca se excluye en este caso por el compromiso renal del paciente. Habría básicamente que diferenciar si es amiloidosis AL o familiar (13). Esta diferencia es importante pues la amiloidosis familiar por transtiretina mutante, que es una proteína de producción hepática, tiene como tratamiento en pacientes seleccionados, transplante hepático, a diferencia de la amiloidosis AL cuyo tratamiento es quimioterapia o transplante de células stem.
Como las técnicas de inmunogold o espectroscopía de masa para amiloide no se realizan en nuestro medio, se hizo inmunohistoquímica en la biopsia de glándula salivar, que resultó positiva para cadenas ligeras lambda en plasmocitos infiltrantes, a la microscopía óptica. Esto sugiere fuertemente amiloidosis AL, siendo un diagnóstico razonable en este paciente que presentaba manifestaciones típicas.
El segundo problema a discutir es si la proteinuria del paciente fue realmente selectiva, tal como fue interpretada inicialmente, dado que la proteinuria de la amiloidosis es típicamente no selectiva.
Dr. Cristian León Rabanal (Nefrología)
Hecho el diagnóstico de amiloidosis sistémica y reevaluado el proteinograma electroforético, es necesario aclarar que la selectividad de proteínas en orina se determina mediante la relación entre el aclaramiento de IgG y el aclaramiento de albúmina o transferrina; considerándose proteinuria selectiva cuando la relación es < 0,2. En este caso, al no contar con estos estudios podemos hacer la interpretación a partir del valor absoluto de gammaglobulinas en orina del paciente (IgG) de 220 mg; siendo los valores normales en orina menores de 3 mg, sugiere que el daño glomerular es tan extenso (secundaria a la enfermedad por depósitos de amiloide), que permite el paso de macromoléculas mayores de 150 000 KDaltons e indicándonos que estamos en realidad frente a una proteinuria no selectiva (14).
Por último nos interesa saber cuál es la terapia actual y el pronóstico de esta condición en general y en este paciente en particular.
Dr. Miro Rodríguez Inocente (Oncología)
El cuadro clínico, la biopsia de glándula salival menor y la inmunohistoquímica (lambda positivo) confirman el diagnóstico de amiloidosis AL. Hay que recordar sin embargo, que en algunos casos éstos están asociados a mieloma múltiple, por lo que hubiera sido interesante completar el estudio de médula ósea y biopsia de hueso. En el 30% de los casos de mieloma se asocian con amiloidosis en la fase tardía de la enfermedad y los del tipo secretor de cadenas ligeras lambda están relacionados a mayor daño renal, a diferencia de los que secretan cadenas ligeras kappa que se asocian más a compromiso óseo e hipercalcemia, por ello es que este paciente no tenía anemia ni lesiones osteolíticas.
El pronóstico de esta entidad es sombrío y el objetivo fundamental del tratamiento es la supresión de la síntesis de la cadena ligera tratando la discrasia de células plasmáticas subyacente con quimioterapia sistémica similar a la terapia del mieloma múltiple siendo melfalán, ciclofosfamida y bortezomib en combinación con dexametasona los más usados. En casos seleccionados, sobre todo en pacientes más jóvenes la quimioterapia a altas dosis con trasplante de células progenitoras (Stem cell) periféricas es la que ha tenido mayor sobrevida con una tasa a 5 años de 60%, pero también es cierto que las tasas de muerte relacionadas al tratamiento es cercana al 10%, que es mucho mayor en relación a los trasplantes realizados por otros desórdenes hematológicos (15). Para finalizar, recordar que en un paciente con amiloidosis se debe buscar mieloma múltiple y a la inversa, en mieloma múltiple hay que buscar amiloidosis sobre todo cuando la paraproteina es a cadenas ligeras lambda.
Editores
La amiloidosis AL y el mieloma múltiple son ambas discrasias de células plasmáticas. Se presentan como entidades separadas pero a veces pueden presentarse concomitantemente, como se ha comentado.
En la amiloidosis AL, el AMO por lo general muestra menos de 10% de células plasmáticas, la cadena ligera es lambda y en pequeñas cantidades (demostrable solo por inmunofijación), no hay hipercalcemia, ni anemia, ni lesiones óseas. La disfunción de órganos es explicada por el depósito de amiloide.
El mieloma múltiple puede ser asintomático o sintomático. El asintomático (que no sería el caso) requiere la presencia de un pico monoclonal de 3 g/dL o de al menos 10% de células plasmáticas en el AMO, en ausencia de daño de órgano blanco. El sintomático requiere daño de órgano blanco (anemia, insuficiencia renal aguda, lesiones óseas líticas o hipercalcemia) y cualquier nivel de paraproteina o de células plasmáticas en médula ósea (generalmente más de 10%). Las cadenas ligeras predominantes son las kappa (16).
Nuestro paciente, al no presentar anemia, azoemia, hipercalcemia o evidencia de lesión ósea y tener cadenas lambda monoclonal y solo manifestarse clínicamente por infiltrado amiloide en corazón y riñón, creemos que el diagnóstico es sólo amiloidosis AL. De haberse realizado la médula ósea y encontrado mas de 10% de células plasmáticas de estirpe monoclonal por inmunofijación, en presencia de daño en órgano blanco, el diagnóstico hubiera sido mieloma múltiple con amiloidosis AL. Sin embargo, dado el mal pronóstico por el compromiso cardiaco severo, la quimioterapia hubiera sido similar a la de amiloidosis AL sin mieloma asociado.
En la evolución posterior, semanas después, el paciente falleció en la sala de hospitalización de otro centro, por edema agudo de pulmón y arritmias letales.
Diagnóstico final
Amiloidosis Sistémica AL
Agradecimientos:
Al Dr. Amador Carcelén Bustamante, por su contribución en la discusión clínica del paciente.
REFERENCIAS BIBLIOGRÁFICAS
1.Kyle RA, Greipp PR. Amyloidosis (AL). Clinical and laboratory features in 229 cases. Mayo Clin Proc. 1983; 58: 665-83. [ Links ]
2.Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. 1997; 337: 898-909. [ Links ]
3.Desai H, Aronow W, Peterson SJ, Frishman WH. Cardiac Amyloidosis: Approaches to diagnosis and management. Cardiol Rev. 2010; 18(1): 1-11. [ Links ]
4.Folk R. Diagnosis and management of cardiac amyloidosis. Circulation. 2005; 112: 2047-2060. [ Links ]
5.Hawkins PM. Hereditary systemic amyloidosis with renal involvement. J Nephrol. 2003; 16: 443. [ Links ]
6.Gillmore JD, Hawkins PN. Drug insight: Emerging therapies for amyloidosis. Nat Clin Pract Nephrol. 2006; 2: 263-270. [ Links ]
7.Gertz MA, Lacy MQ, Dispenzier A. Immunoglobulin light chain amyloidosis and the kidney. Kidney Int. 2002; 61:1-9. [ Links ]
8.Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006; 354 (13):1362-9. [ Links ]
9.Chee CE, Lacy MQ, Dogan A, Zeldenrust SR. Pitfalls in the diagnosis of primary amyloidosis. Clin Lymphoma Myeloma Leuk. 2010; 10 (3):177-80. [ Links ]
10.Gertz MA. How to manage primary amyloidosis. Leukemia. 2011; 1-8. URL disponible en http://www.ncbi.nlm.nih.gov/pubmed/21869840. (Fecha de acceso: diciembre 2011) [ Links ]
11.Arbustini E, Morbini P, Verga L, et al. Light and electron microscopy immunohistochemical characterisation of amyloid deposits. Amyloid. 1997; 4: 157-170. [ Links ]
12.Connors L, Davidoff R, Skinner M, Falk R. Senile systemic amyloidosis presenting with heart failure: a comparison with light chain-associated amyloidosis. Arch Intern Med. 2005; 165:1425-1429. [ Links ]
13. Di Girolamo M, Monno D, Pirro MR, Nowakowski M. Approach to diagnosis in systemic amyloidosis: initial findings and time from symptoms onset to diagnosis (light-chain amyloidosis vs. transthyretin). Problems and observations. Amyloid. 2011; 18 ( S1): 83-85. [ Links ]
14.Carlson JA, Harrington JT. Laboratory evaluation of renal function. En: Schrier RW, Gottschalk CW. (Editors). Diseases of the kidney. Boston: Little Brown and Company. 1993. p. 361–405. [ Links ]
15.Rajmur SV, Gertz MA. Advances in the treatment of Amyloidosis. New Engl J Med. 2007; 356: 2413-2415. [ Links ]
16.International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003; 121:749-57. [ Links ]
