











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkConversatorio clínico-patológico en el Hospital Nacional Arzobispo Loayza 2012-2. Caso 02-2012. Mujer de 44 años de edad, con tos productiva, disnea y hematuria con sedimento urinario telescopado.
Clinical case at the Hospital Nacional Arzobispo Loayza. Case 02-2012. Female 44 years old with productive cough, dyspnea and hematuria with telescoped urinary sediment.
Edmundo Alegría1, Marco J. García2, César Chian3.
1 Médico residente de tercer año en Medicina Interna. Facultad de Medicina Alberto Hurtado. Universidad Peruana Cayetano Heredia. Lima, Perú.
2 Médico Especialista en Medicina Interna, Hospital Nacional Arzobispo Loayza. Lima, Perú.
3 Médico asistente. Dpto. de Anatomía Patológica. Hospital Nacional Arzobispo Loayza. Profesor Asociado. Dpto. de Patología. Facultad de Medicina Alberto Hurtado. Universidad Peruana Cayetano Heredia. Lima, Perú.
PALABRAS CLAVE: Vasculitis, Granulomatosis de Wegener.
KEYWORDS: Vasculitis, Wegener Granulomatosis.
Enfermedad Actual
Mujer de 44 años, casada, ama de casa, natural de La Oroya y procedente de Jauja. Historia de 6 meses de tos poco productiva, de leve intensidad que luego se hizo productiva con expectoración blanquecina; además, presenta sensación nocturna de alza térmica.
Un mes antes, la expectoración se vuelve hemoptoica, por lo cual fue llevada otro hospital donde le diagnostican Quiste Hidatídico derecho infectado. Además, presentaba disfonía progresiva. Dos semanas antes del ingreso, se agrega hipoacusia bilateral y disnea. Cuatro días antes del ingreso presentó lesiones equimóticas en miembros inferiores y codos, y tres días antes del ingreso aparecieron lesiones aftosas en la boca; por este motivo acudió a nuestro hospital.
Funciones biológicas: Apetito disminuido; sed conservada; orina disminuida; deposiciones 1 vez/día y sueño alterado.
Antecedentes
Diagnóstico de Quiste Hidatídico en marzo 2010, en el Hospital de Huancayo. Cesárea hace aproximadamente 2,5 años, por parto pre-termino.
G8 P8008, FUR: Hace aproximadamente 2 años. Antecedentes familiares: No contributorios.
Examen Físico
PA: 110/60; FC: 84x; FR: 20x; Temperatura: 36,6°C.
Paciente despierta, en regular estado de nutrición. Piel pálida, con lesiones equimóticas, vesiculares y flictenulares en los cuatro miembros. Edema en los cuatro miembros. Cuello: no había bocio. Pulmones: murmullo vesicular disminuido en bases pulmonares; roncantes difusos y algunos sibilantes en ambos campos pulmonares. Aparato cardiovascular: Ruidos cardiacos rítmicos de buena intensidad, no había ingurgitación yugular. Abdomen y genito-urinario, normal. Neurológico: despierta, pares craneales, reflejos osteotendinosos, sensibilidad, coordinación: normales.
Los exámenes hematológicos y bioquímicos se muestran en la tabla 1. El examen de orina mostró proteínas 3+, sangre 3+, glucosa 1+, leucocitos: 11-20 x c, hematíes >100 x c; cultivo de secreción bronquial: Staphylococus aureus. VDRL: No reactivo; VIH: no reactivo; Anticuerpos antinucleares: negativos; ANCA-P: positivo, Grupo sanguíneo B+
Evolución
Al segundo día de hospitalización la paciente presentó disnea, la que fue aumentando progresivamente y se agregó oliguria severa. Al tercer día, presentó somnolienta, hipotensión, mayor dificultad respiratoria, y continuó con oliguria, lo que motivó su ingresó a la unidad de cuidados intensivos para soporte hemodinámico y ventilatorio.
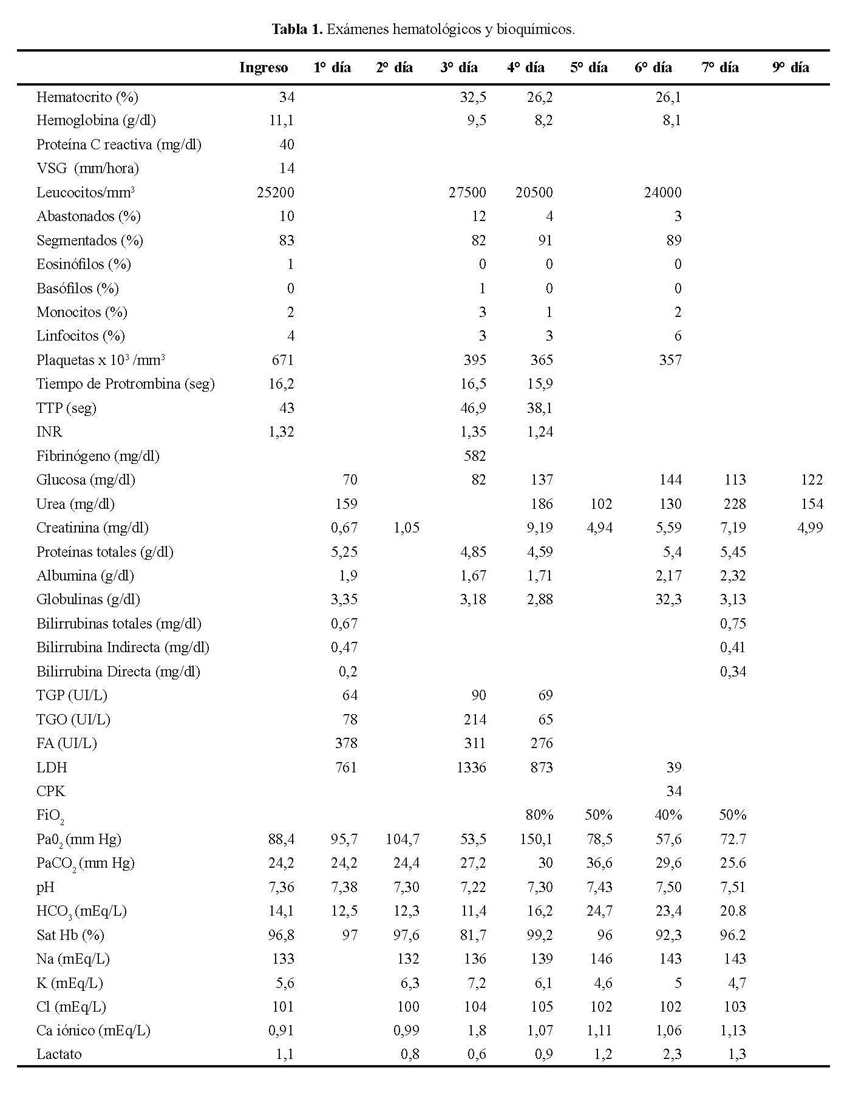
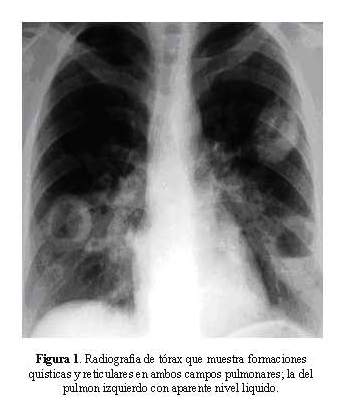
La radiografía de tórax mostró formaciones quísticas y reticulares en ambos campos pulmonares; la del pulmón izquierdo con aparente nivel liquido ( Figura 1). La tomografía de tórax mostró lesión de masa en el hemitórax derecho con necrosis en el centro y bordes espiculados, además se observan lesiones micronodulares en ambos campos pulmonares ( Figura 2).El PAP de aspirado bronquial mostró macrófagos cargados de pigmento.
Discusión Clínica:
Dr. Marco Julio García
Debemos identificar los problemas que tiene el paciente. El primer problema es el compromiso pulmonar con disnea progresiva, asociada a expectoración hemoptoica y evidencia de lesión parenquimal pulmonar en la radiografía de tórax.
El siguiente problema, es el compromiso renal; en el examen de orina hay hematuria con sedimento urinario telescopado. También tenía compromiso dérmico con púrpura palpable, con lesiones equimóticas; además trastorno del medio interno evidenciándose una acidosis metabólica bien marcada con hipoalbuminemia.
Nos enfrentamos a un problema sistémico. Las preguntas a responder son: 1) ¿Tiene una hemorragia alveolar?, 2) ¿Es un proceso infeccioso que está produciendo expectoración hemoptoica?, y 3) ¿El sedimento urinario será suficiente para hacer un diagnóstico etiológico de la lesión renal?
Respondiendo las preguntas:
- En la hemorragia alveolar hay macrófagos cargados con hemosiderina en el esputo o en el lavado bronquial; paralelamente, caída del hematocrito. También se establece el diagnostico por histopatología, esta muestra lesiones granulomatosas o vasculitis en el 90 % de los casos (1), pero es riesgoso realizar una biopsia de pulmón en presencia de una inflamación capilar.
En la radiografía de tórax, la hemorragia alveolar, principalmente la difusa, se acompaña de un infiltrado alveolar difuso y bilateral.
- Si, hay agentes patógenos que pueden manifestarse con hemoptisis, todos ellos existen en el Perú: Tuberculosis, Pneumocistis carini en pacientes inmunocomprometidos, Strongiloidiasis, Legionella.
- La historia clínica nos permite hacer el diagnóstico de glomerulonefritis rápidamente progresiva, con trastorno hemodinámico, azoemia, con evolución tórpida menor de tres meses, que asociadas a la hemorragia alveolar difusa constituyen una forma del síndrome pulmón-riñón.
Existen varias formas de este síndrome, pueden ser: vasculitis primarias o secundaria (2). Las primarias comprometen vasos muy pequeños, son la poliangeitis microscópica, la enfermedad de Wegener y el síndrome de Churg-Strauss, y están asociados al ANCA (3); por otro lado, el síndrome de Goodpasture, que tiene anticuerpos anti membrana basal; el Lupus eritematoso sistémico, puede complicar el diagnóstico.
En la enfermedad de Wegener la lesión pulmonar es común (85%); en la poliangeitis microscópica es bastante menor y es inusual en la enfermedad de Churg-Strauss (1). Las lesiones en el tracto aéreo superior, ocurren en el 92% de los casos (1) y la paciente tiene lesiones ulceradas en el tracto respiratorio superior, que es frecuente en la enfermedad de Wegener.
Otra evidencia, es la positividad del ANCA P, que ocurre en casi todos los pacientes con enfermedad de Wegener, particularmente cuando está activa (3).
La lesión pulmonar en la enfermedad de Wegener se caracteriza por una alveolitis difusa con infiltración de neutrófilos; esto da un pronóstico malo en relación con casos de infiltración linfocitaria, en los cuales el pronóstico es mejor. La hemorragia alveolar extensa causada por capilaritis tiene una mortalidad de 50% (4).
En otras entidades también se pueden encontrar neutrófilos cargados con hemosiderina, tales como la insuficiencia cardiaca prolongada, infecciones pulmonares como tuberculosis, por Pnemocistis carini, aspergillosis, etc.
En la radiografía de tórax de estos pacientes se puede encontrar: imágenes nodulares, a veces cavitadas simulando un absceso, en otros casos, infiltrados difusos o derrame pleural (1).
ANCA es la abreviatura de anticuerpos anti citoplasmáticos; anticuerpos que muestran una distribución granular difusa por inmunofluorescencia; la variedad C corresponde a aquellos dirigidos contra la proteinasa 3 y es la más importante en esta enfermedad, en tanto que, los ANCA P, tinción perinuclear, están dirigidos contra la mieloperoxidasa y su presencia no es útil para el diagnóstico de enfermedad de Wegener (1).
La vasculitis asociada a ANCA y no tratada, tiene una mortalidad del 90% en dos años, principalmente por complicaciones pulmonares y con un tratamiento adecuado usando inmunosupresores y corticoides, la remisión puede llegar a 93% a 4 años (5).
Diagnóstico Clínico: Enfermedad de Wegener.
Discusión Patológica
Dr. César Chian
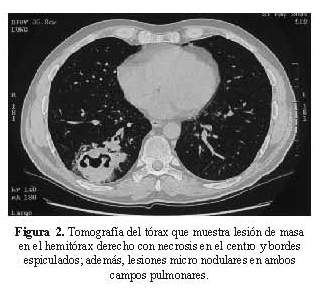
Se recibió una biopsia de piel que al examen microscópico presentaba ulceración, extensa necrosis epidérmica, tejido de granulación, edema de la dermis y a nivel de vasos de la dermis intermedia y profunda, grandes trombos de fibrina ocluyendo la luz vascular, con presencia de infiltrado inflamatorio en la pared de los vasos sanguíneos, necrosis fibrinoide del endotelio y cariorrexis ( Figura 3 y figura 4). En los cortes histológicos no se observaron granulomas.
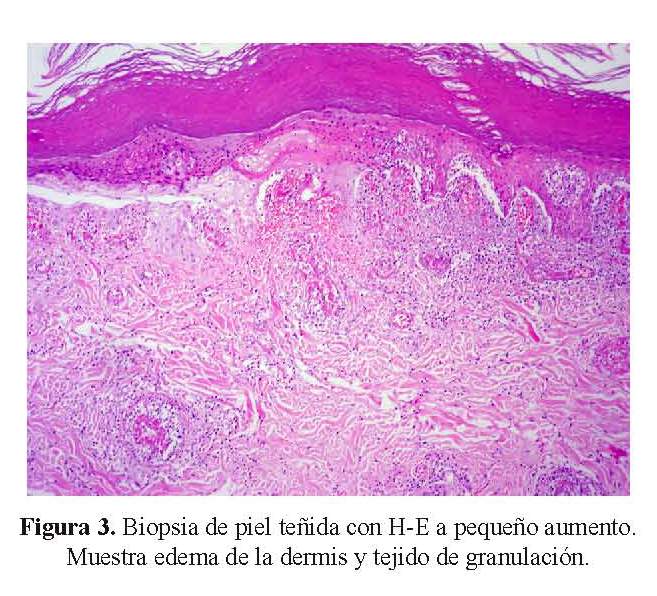
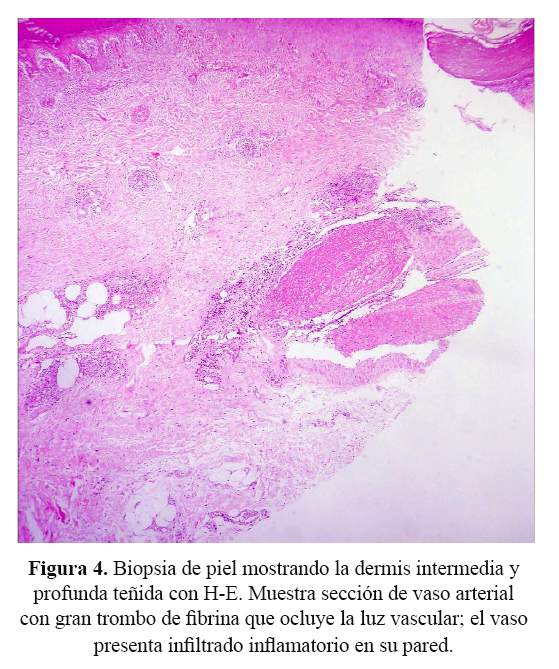
Los hallazgos histológicos corresponden a una vasculitis de vasos medianos y pequeños con leucocitoclasia. Los criterios histológicos para el diagnóstico de vasculitis se clasifican en mayores y menores (6). Los criterios mayores incluyen: 1) presencia de neutrófilos y cariorrexis (polvo nuclear) en las paredes de los vasos sanguíneos, y 2) necrosis de células endoteliales y de las paredes vasculares con depósitos de fibrina. Los criterios menores incluyen: Edema endotelial, extravasación de glóbulos rojos, trombosis, necrosis epidérmica, vesículas epidérmicas y dérmicas, fibroplasia reparativa perivascular, dilataciones aneurismáticas y necrosis de glándulas anexas.
En nuestro caso los dos criterios mayores estaban presentes y también varios de los criterios menores.
Los sistemas de clasificación utilizados en vasculitis sistèmicas son diversos, algunos se basan en la patogénesis y dividen a las vasculitis en mediadas por ANCA y mediadas por complejos inmunes; dando síndromes clínicos agrupados según la evolución o respuesta a la terapia (7).
Actualmente los dos sistemas más utilizados son los de Chapell Hill y del Colegio Americano de Reumatología, que, primero categorizan a las vasculitis según el tamaño del vaso y a partir de allí configuran entidades clínico patológicas con características distintivas (8). Las entidades incluidas entre las vasculitis de vasos pequeños y medianos en estas dos clasificaciones son: poliarteritis nodosa y enfermedad de Kawasaki (vasos medianos) y granulomatosis de Wegener, enfermedad de Churg-Strauss, poliangeitis microscópica, púrpura de Henoch-Schönlein, crioglobulinemia esencial y angeitis leucocitoclástica cutánea (vasos pequeños).
Desde el punto de vista patológico no existen criterios, (más allá del tamaño del vaso afectado), que sean indicativos de la etiología o diagnóstico definitivo del tipo de vasculitis. En algunos casos como en la poliarteritis nodosa en que la afectación de los vasos no es uniforme en toda la longitud, sino en forma saltatoria (de allí el nombre de nodosa), o en la vasculitis de Churg-Strauss que suele presentar una gran cantidad de eosinófilos, no se puede sugerir un diagnóstico específico, siendo indispensable el correlato clínico y el uso de pruebas de laboratorio para el diagnóstico definitivo.
Desde el punto de vista etiológico, es importante considerar estudios que indican que aproximadamente el 40% de vasculitis cutáneas son idiopáticas, 10% corresponden a púrpura de Henoch-Schoenlein, menos de 5% están asociadas a vasculitis sistémica primaria, alrededor de 10% corresponden a enfermedades del tejido conectivo y un importante 20% está relacionada a infecciones (7).
Haciendo unidad con la discusión clínica podemos decir que las manifestaciones cutáneas de vasculitis que observamos corresponden a vasos medianos y pequeños, siendo nuestros diagnósticos diferenciales de granulomatosis de Wegener, poliarteritis nodosa y poliangiitis microscópica. La prueba de mayor valor para hacer el diagnóstico diferencial entre estas entidades es la determinación de anticuerpos antineutrófilos. Es importante destacar que aproximadamente el 40% de pacientes con granulomatosis de Wegener, mientras el 50% de pacientes con PAN o poliangiitis microscópica presentan un compromiso cutáneo (3). Pese a que, la presencia de inflamación extravascular con granulomas a nivel intersticial, se considera característica de granulomatosis de Wegener, la ausencia de granulomas no excluye esta posibilidad diagnóstica.
Diagnóstico patológico: Enfermedad de Wegener.
Diagnóstico Final
Enfermedad de Wegener
REFERENCIAS BIBLIOGRÁFICAS
1.Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener Granulomatosis: An analysis of 158 patients. Ann Intern Med. 1992; 116 (6): 488-98. [ Links ]
2.Watts RA, Scott DG. Classification and epidemiology of the vasculitides. Balliere´s Clinical Rheumatology. 1997; 11(2):191-217. [ Links ]
3.Moosig F, Lamprecht P, Gross WL. Wegeners´s Granulomatosis: The current view. Clinic Rev Allerg Immunol. 2008; 35:19-21. [ Links ]
4.Manganelli P, Fietta P, Carotti M, Pesci A, Salaffi F. Respiratory system involvement in systemic vasculitis. Clin Exp Rheumatol. 2006; 24: S48-59. [ Links ]
5.Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener´s Granulomatosis: Prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med. 1983; 98:76-85. [ Links ]
6.Farmer E, Hood A. Pathology of the Skin. 2nd Ed. New York: McGraw-Hill; 1999. p. 4-8. [ Links ]
7.Carlson JA, Ng BT, Chen KR. Cutaneous vasculitis update: diagnostic criteria, classification, epidemiology, etiology, pathogenesis, evaluation and prognosis. Am J Dermatopathol. 2005; 27(6):504-528. [ Links ]
8.Florentino D. Cutaneous vasculitis. J Am Acad Dermatol. 2003; 48:311-40. [ Links ]
