











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkGran Ronda de Medicina Interna y Especialidades del Hospital Nacional Cayetano Heredia / Caso clínico 02-2012. Varón de 38 años con neuropatía periférica, edemas y lesiones dérmicas.
Grand Round of Internal Medicine and specialties at the Hospital Nacional Cayetano Heredia./ Clinical case 02-2012. A 38 years-old man with peripheral neuropathy, edema and dermatologic lesions.
Editor de sección: Dr. Sergio Vásquez Kunze; Editores asociados: Dr. Héctor Sosa Valle, Dr. Ray Ticse Aguirre, Dr. Leslie Soto Arquiñigo.
Liliana Cabrera 1, Sergio Vásquez 2, Alejandro Escalaya 3, César Ramos 4, Francisco Bravo4, Diego Venegas 5.
1Médico Residente de 3° año de Medicina Interna. Dpto. de Medicina, Hospital Nacional Cayetano Heredia. Lima, Perú.
2Médico asistente, Servicio de Medicina Interna. Dpto. de Medicina, Hospital Nacional Cayetano Heredia. Lima, Perú.
3Médico asistente Servicio de Neurología. Dpto. de Medicina, Hospital Nacional Cayetano Heredia. Lima, Perú.
4Médico asistente, Servicio de Dermatología. Dpto. de Medicina, Hospital Nacional Cayetano Heredia. Lima, Perú.
5Médico asistente, Servicio de Hemato-Oncología. Dpto. de Medicina, Hospital Nacional Cayetano Heredia. Lima, Perú.
CASO CLÍNICO
Varón de 38 años con debilidad en extremidades, edema de miembros inferiores y lesión dérmica violácea eritematosa en región pectoral de 1 año de evolución. El paciente inició sus síntomas hace un año, con parestesias y debilidad de extremidades. La debilidad era a predominio de piernas, bilateral, simétrico y de progresión ascendente; fue empeorando rápidamente y desde 6 meses antes de su admisión la impedía la deambulación. No presentaba otros síntomas de focalización.
Por este motivo acudió a un instituto neurológico especializado donde se le realizaron diversos estudios, encontrándosele glucosa 240 mg/dl, TSH 6,3 µU/ml (VN 0,27- 4,2uU/ml), T4 libre 1,05 µg/ml (VN 0,89 -1,76 µg/ml), plomo sérico 21,3 µg/dl (VN menor a 20 µg/dl) y un estudio de velocidad de conducción nerviosa (VCN) compatible con polineuropatía axonal distal a predominio de miembros inferiores. El hemograma, las pruebas de función renal y hepática, electrólitos, proteinograma electroforético sérico y examen completo de orina fueron normales. Por estos hallazgos se le diagnosticó polineuropatía plúmbica, hipotiroidismo subclínico y diabetes mellitus, indicándosele carbamazepina 200 mg cada 12h, prednisona 70 mg diarios que fue disminuyéndose progresivamente hasta retirarse 3 semanas después, D-penicilamina 250 mg cada 8h, levotiroxina 125 µg al día y metformina 850 mg diarios. Con el tratamiento presentó mejoría parcial de la debilidad, sin llegar a deambular. El control de plomo sérico al mes de tratamiento fue 1,3 µg/dl.
Cinco meses antes del ingreso la debilidad empeoró, y presentó además edema en miembros inferiores y la aparición progresiva de una lesión eritemato-violácea en la región pectoral de aproximadamente 20x15 cm que evolucionó a una placa descamativa ( Figura 1).
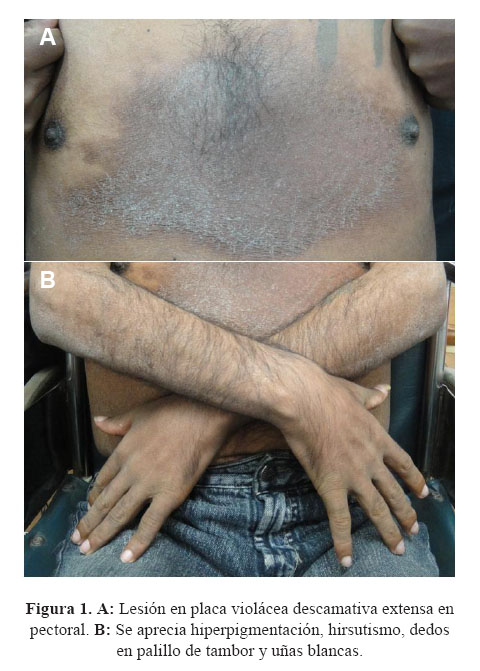
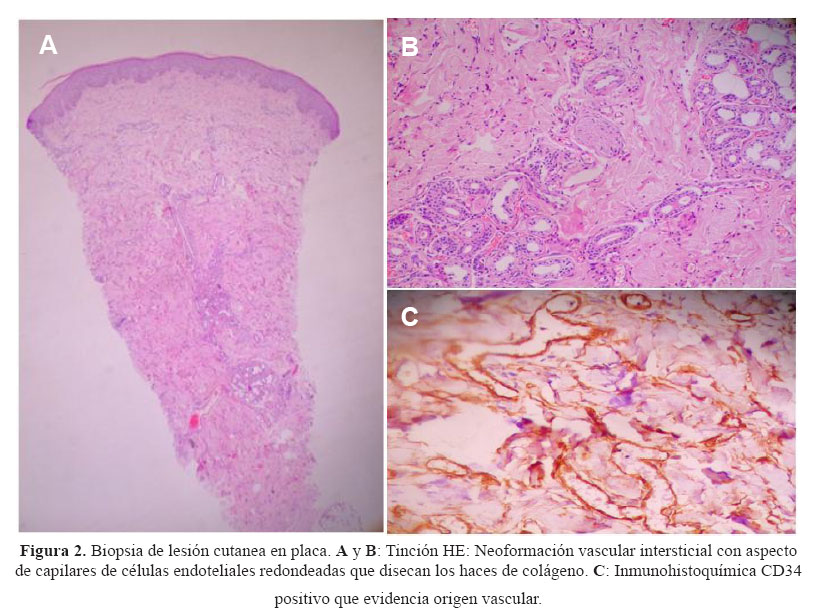
Cuatro meses antes del ingreso el paciente no podía deambular sin uso de muletas y poco tiempo después solo se movilizaba en silla de ruedas. Acudió a nuestra institución siendo evaluado ambulatoriamente en los servicios de neurología, medicina interna y dermatología. En este último se le realizó una biopsia de la placa eritemato-violácea de bordes irregulares en la región pectoral, que fue informada como una neoformación vascular intersticial con aspecto de capilares de células endoteliales redondeadas que disecaban haces de colágeno. La inmunohistooquimica fue positiva para CD 34, que confirmaba su origen vascular, y ante la sospecha de Sarcoma de Kaposi se realizó una tinción para herpes virus 8, que fue negativa ( Figura 2).
En la evaluación neurológica se encontró la fuerza en miembros superiores proximal 5+/5, distal 4+/5, en miembros inferiores proximal 3+/5, distal 3+/5; hiporeflexia generalizada, hipotonía en miembros inferiores, Lasegue leve bilateral y alteración de la vibración. El fondo de ojo mostró edema de papila bilateral.
Se le realizó una nueva electromiografía y velocidad de conducción siendo compatible con una polirradiculoneuropatía sensitivo motora crónica de tipo desmielinizante. En la punción lumbar se obtuvo un LCR cristal de roca, presión de apertura 14 cm de agua, con proteínas 165 mg/dl, glucosa 60 mg/dl y ausencia de células. Se le realizaron nuevos exámenes auxiliares que mostraron hemoglobina en 19,2 g/dl, hemograma con leucocitos 12 000/ mm3 con fórmula normal, plaquetas 372 000/mm3, VSG 2 mm/h, glucosa 103 mg/dL, proteínas en 5,8 g/dl y albúmina 3 g/dl. Las pruebas de función hepática, renal y examen de orina fueron normales. La inmunofijación sérica mostró elevación de Ig A, Ig G y de cadenas kappa y lambda, siendo informada como gammapatia policlonal.
Se le realizó un aspirado de médula ósea que reveló celularidad incrementada para la edad del paciente, maduración adecuada de las 3 series, hiperplasia severa de la serie megacariocitica, presencia de células plasmáticas en 16% algunas de ellas atípicas y hierro medular ausente; en la biopsia de hueso se encontró coágulo y hueso con celularidad de 20-30% con respecto al tejido adiposo y presencia de las tres series hematopoyéticas con buena maduración, eosinofilia leve e incremento de las células plasmáticas menor al 10%; no se observaron granulomas ni células de aspecto neoplásico.
Un mes antes del ingreso se le indicó dexametasona 8 mg cada 8 horas en 3 pulsos de 4 días cada uno sin presentar mejoría clínica, motivo por el que se decidió su hospitalización: Tenía hiporexia, había perdido 17 kg en 5 meses y la orina y deposiciones no presentaban alteraciones.
El paciente negaba historia de hipertensión arterial, tuberculosis, fiebre malta, infección por hepatitis B o C, no tenía contactos con tuberculosis, ni transfusiones previas. Fue operado de apendicetomía hace 10 años. Consumía moderadamente cerveza una vez al mes y no fumaba. El paciente era técnico de sistemas de aire acondicionado y estuvo expuesto a gases y pinturas industriales. Además trabajó en minería por 3 meses hace 2 años. El paciente mantenía una relación monógama desde hace 10 años y no refería conducta sexual de riesgo para enfermedades transmisibles. Su padre tenía diabetes mellitus tipo 2 y falleció de cáncer de colon.
La revisión anamnésica de sistemas y aparatos reveló disminución leve de la agudeza visual, hipertricosis en pecho, brazos y piernas; hiperpigmentación en piel y mucosas, y disfunción eréctil de reciente aparición.
Al examen físico tenía presión arterial 110/80 mm Hg de pie y 100/70 mm Hg en decúbito, pulso 89 por minuto, frecuencia respiratoria 22 por minuto y estaba afebril. Lucía en mal estado general, desnutrido y se movilizaba en sillas de ruedas. La piel era seca, descamativa y presentaba hiperpigmentación generalizada que incluía mucosas, además una placa eritematosa de aproximadamente 20x15 cm en la región pectoral; tenía hipertricosis, uñas blancas y presencia de dedos en palillo de tambor (Figura 1:B). El tejido celular subcutáneo estaba disminuido y tenía edema 2+/3+ con fóvea en miembros inferiores. Se palpaban múltiples ganglios cervicales de aproximadamente 2x2 cm, móviles e indoloros. No había anormalidades en los pulmones ni en el aparato cardiovascular. El abdomen era blando y sin dolor, y el span hepático en 15 cm. El examen neurológico mostró un paciente despierto, lúcido en las tres esferas, con disminución de la fuerza muscular (3/5) proximal y distal en piernas y 4/5 proximal y distal en brazos; tenía hipotrofia marcada, flacidez bilateral e hiporeflexia generalizada; la sensibilidad estaba conservada, excepto por la vibración que estaba disminuida y en el fondo de ojo se encontró papiledema bilateral. El resto de la evaluación fue normal.
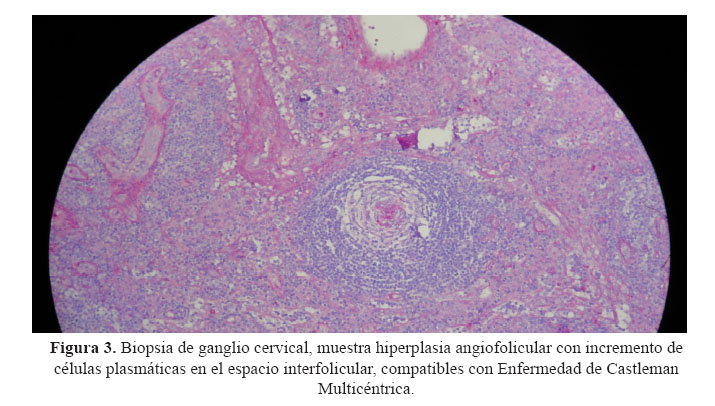
Se realizaron exámenes complementarios que incluyeron una radiografía de tórax que no mostró alteraciones significativas y radiografías de cráneo, parrilla costal, columna vertebral, pelvis ósea y extremidades en las que no se apreciaron lesiones. La determinación de cadenas ligeras en orina de 24 horas fue negativa. La biopsia de ganglio cervical mostró hiperplasia angiofolicular con incremento de células plasmáticas en el espacio interfolicular ( Figura 3).
DISCUSIÓN
La discusión se centró en el diagnóstico etiológico y sobre otro procedimiento clave o examen auxiliar para llegara al diagnóstico. Se invitaron a la presentación a los médicos de los servicios de medicina interna, neurología, dermatología y hemato-oncología.
Dr. Sergio Vásquez Kunze (Medicina Interna)
Este paciente presenta 3 problemas cardinales, la polineuropatía (PN) desmielinizante, que me parece el principal, las lesiones dérmicas y el edema.
La más común de las neuropatías es la diabética, pero ésta generalmente es axonal. Al paciente se le diagnosticó diabetes, pero una neuropatía severa como esta no es común en el debut. Es un diagnóstico asociado pero no impresiona ser la causa.
El diagnóstico inicial de PN por exposición crónica a plomo que derivó en tratamiento, es muy cuestionable. Si bien el paciente pudo tener una exposición crónica y el nivel de plomo sanguíneo ligeramente elevado, éste no se considera significativo para una PN agresiva como la que tenía. Aun niveles entre 30 y 70 ug/dL pueden no presentar síntomas o ser muy inespecíficos como mialgias, fatiga, irritabilidad, insomnio, anorexia y alteraciones en la memoria. La corrección posterior no mejoró los síntomas. La intoxicación por plomo produce una PN axonal, tal vez la primera VCN influyó en ese diagnóstico.
El hipotiroidismo hallado inicialmente es una causa rara de neuropatía. Parece también una enfermedad asociada.
El diagnóstico diferencial de PN desmielinizante es limitado: las genéticas como el síndrome de Charcot Marie Tooth 1 se descartan porque son crónicas, de muchos años. Dentro de las inflamatorias, la PN inflamatoria crónica desmielinizante es la mas frecuente, similar a un Síndrome de Guillain Barre pero de curso crónico, responden a esteroides o inmunoglobulina, sin embargo, no tiene las características sistémicas de nuestro paciente. Las asociadas a enfermedades del colágeno como lupus o Sjogren, tóxicas como difteria, farmacológicas (amiodarona), paraneoplásicas (linfoma) e infecciosas (VIH y Lyme) no se ajustan a la historia y antecedentes de nuestro paciente. Por último están las asociadas a paraproteinas (cadenas ligeras lambda o kappa), como el mieloma múltiple (MM), gamapatía monoconal de significado incierto (GMSI), macroglobulinemia de Waldenstrom (MW), amiloidosis AL y el síndrome de POEMS.
Aunque no se ha demostrado una paraproteina monoconal en este paciente es interesante comentarlo. Los plasmocitos secretan cadenas ligeras (por lo general lambda) en lesiones desmielinizantes y estas atacan los GM1, sulfatides o la glicoproteína asociada a mielina. La GMSI es la más frecuente, pero se ve en personas mayores. El paciente tampoco tiene clínica de MM (anemia, falla renal, lesiones osteolíticas). La MW tiene adenopatías con hallazgos de linfoma e hiperviscosidad, tampoco presentes en este paciente. La amiloidosis AL produce mas una PN axonal y no explica los cambios dérmicos. Por último está el síndrome de POEMS (polineuropatía, organomegalia, alteraciones endocrinológicas, gamapatia monoclonal y alteraciones dérmicas). Dado que nuestro paciente tiene lesiones dérmicas proteicas como segundo problema cardinal y alteraciones endocrinológicas, es un diagnóstico a discutir en detalle.
El paciente tiene además otras manifestaciones diversas no explicadas como edema prominente sin causa aparente, disfunción eréctil, papiledema sin hipertensión endocraneana, policitemia y trombocitosis, leve hepatomegalia, clubbing, adenopatía cervical e incremento de las células plasmáticas en medula ósea. ¿Pueden todas estas manifestaciones corresponder a un síndrome de POEMS también?
¿Tiene este paciente un síndrome de POEMS? Éste un síndrome paraneoplásico secundario a neoplasia de células plasmáticas, generalmente mieloma osteoesclerótico, no mieloma múltiple. Tiene mayor incidencia en la raza asiática, en el sexo masculino (2,5:1), y la edad de presentación es entre los 40 y 50 años (1).
Aparte de las características clásicas que definen su acrónimo tiene muchas otras que han sido agrupadas en orden de importancia y frecuencia en los criterios de la Clínica Mayo (2). Muchos de estos están presentes en nuestro paciente. La polineuropatía y el pico monoclonal son los 2 criterios mayores y mandatorios. Luego están las lesiones escleróticas, enfermedad de Castleman y los niveles aumentados del factor de crecimiento endotelial vascular que son también mayores pero no mandatorios, y luego están varias características calificadas como menores: la organomegalia (esplenomegalia, hepatomegalia o linfadenopatía), sobrecarga de volumen extravascular (edema, efusión pleural o ascitis), endocrinopatía (adrenal, tiroidea, pituitaria, gonadal, paratiroidea o pancreática), cambios cutáneos (hiperpigmentación, hipertricosis, hemangioma glomerular, uñas blancas, acrocianosis), papiledema y policitemia o trombocitosis.
El diagnóstico de POEMS es confirmado si tiene 2 criterios mayores mandatorios, un criterio mayor no mandatorio y uno de los 6 criterios menores.
Actualmente se reconoce que la causa de las manifestaciones del Síndrome POEMS no son bien conocidas y la cadena lambda monoclonal no participa en la mayoría de manifestaciones, excepto en la PN. En cambio parece importante ahora en la fisiopatología la producción de citoquinas proinflamatorias como el factor vascular de crecimiento endotelial (FVCE), IL6, IL1B, FNT alfa (3). El aumento de la permeabilidad vascular por el aumento del FVCE se reconoce como la causa del edema, la ascitis, la organomegalia, neovascularización, papiledema y probablemente la PN (4). Las células plasmáticas son una fuente mayor del FVCE en este síndrome (5).
¿Cumple nuestro paciente con los criterios para POEMS definido? De los criterios mayores mandatorios tiene la PN desmielinizante, pero la presencia de pico monoclonal (lambda) esta ausente. El paciente fue estudiado por inmunofijación (IF) en sangre y orina, que es método más sensible para hallar pico monoclonal. El proteinograma electroforético es menos sensible y es normal hasta en el 25% de pacientes. Sin embargo, cuando se estudia por IF la sensibilidad llega hasta 96,8% (6). El 3% restante puede ser detectado por inmunohistooquimica (IH) o citometría de flujo (CF) en la médula ósea. También se pueden cuantificar las cadenas libres lambda y kappa en sangre y se hace un cociente lambda/kappa, si éste es elevado también es expresión de monoclonalidad (7).
Este paciente tuvo policlonalidad en la IF, pero por lo antedicho acerca de la dificultad en escasos pacientes de identificar el pico monoclonal no se puede rechazar completamente un POEMS, pero tampoco se puede diagnosticar categóricamente.
En los criterios mayores no mandatorios, se describe la enfermedad de Castleman. Nuestro paciente presentó hiperplasia angiofolicular con incremento de células plasmáticas en el espacio interfolicular en la biopsia de ganglio. Estos hallazgos son compatibles con enfermedad de Castleman multicéntrica (ECM). Esta se asocia entre 19 - 24% al síndrome de POEMS (1). El 80% de los pacientes que presentan esta asociación son positivos a HHV-8, que está fuertemente asociado a ECM. Es de notar que la etiopatogenia de ECM esta ligada también al aumento de la IL6 (8).
Otro criterio mayor es el mieloma osteoesclerótico (MOS). El paciente no lo presentó en las radiografías convencionales. En la serie de la Clínica Mayo se describe que bajo este método se identifica una lesión ósea en el 97% de pacientes (1). Sin embargo, recientemente se han descrito casos con radiografías negativas y tomografía por emisión de positrones (PET) positiva, los MOS detectados por ésta técnica suelen ser lesiones osteoescleroticas muy pequeñas y en sitios técnicamente difíciles como el sacro (9). No se le realizó éste estudio (PET) por motivos económicos; tampoco se le realizó FCVE.
De los criterios menores en este paciente destaca la lesión dérmica tipo placa. La hiperpigmentación e hipertricosis se puede ver en el 50 y 25% respectivamente (1).
Si bien tiene otros muchos criterios menores hay que mencionar que ni la diabetes ni el hipotiroidismo forman parte de los criterios endocrinológicos por su elevada frecuencia en la población general. Es de notar que el hipogonadismo es la anormalidad endocrinológica mas frecuente y hasta el 70% de varones tiene disfunción eréctil (1), como lo presentaba nuestro paciente. Hubiera sido interesante el dosaje de testosterona para confirmarlo.
En resumen, el paciente no tiene pico monoclonal ni lesión osteoesclerótico pero tiene un fenotipo de POEMS característico. ¿Podría haber otra etiología para esta presentación?
La relación entre la enfermedad de Castleman y Síndrome POEMS. La enfermedad de Castleman es una enfermedad linfoproliferativa que se caracteriza por adenopatías y síntomas sistémicos tipo B. Hay 2 variedades histopatológicas la hialina-vascular (generalmente solitaria) y la plasmática (generalmente multicéntrica), esta última fue la que presentó nuestro paciente. Ambas tienen riesgo de evolución a linfoma.
La variedad plasmática puede asociarse a síndrome POEMS. Actualmente se reconoce que ambas condiciones pertenecerían al espectro de enfermedad por células plasmáticas que producen FCEV e IL6 (2). Es de notar que cuando la enfermedad Castleman se asocia a síndrome POEMS puede presentar gamapatía policlonal y ausencia de mieloma osteoesclerótico, como es el caso de nuestro paciente. En ese caso se le ha denominado enfermedad de Castleman variante de POEMS para diferenciarla del Síndrome POEMS clásico con hallazgos de enfermedad de Castleman en sus adenopatías y que si presentan gamapatía monoclonal y MOS (2).
Bajo estas perspectivas los 2 mejores diagnósticos de este paciente son: Síndrome de POEMS sin pico monoclonal ni mieloma osteoesclerótcio encontrado por las técnicas usadas, o Enfermedad de Castleman variante de POEMS (policlonal y sin MOS). Creo que el paciente debe recibir tratamiento agresivo como un Síndrome POEMS o una Enfermedad de Castleman variante POEMS, es decir una discrasia de células plasmáticas.
Dr. Alejandro Escalaya Advíncula (Neurología)
Estamos ante un paciente con un cuadro crónico caracterizado por síntomas motores y sensitivos a predominio de miembros inferiores. La ausencia de hiperreflexia, nivel sensitivo y signo de Babinski positivo alejan la posibilidad de una mielopatía y orientan hacia un desorden del sistema nervioso periférico. El patrón de debilidad próximo-distal y el signo de Lasegue bilateral ayudan clínicamente a localizar la afección a nivel poliradicular. El hallazgo de percepción de la vibración disminuida traduce además una polineuropatía de fibras largas (10). Las fibras largas están densamente mielinizadas y, por consiguiente, se comprometen en procesos básicamente desmielinizantes. La presencia de parestesias (síntoma sensorial positivo) es un hallazgo útil porque muchas neuropatías adquiridas están acompañadas de síntomas sensoriales neuropáticos positivos y muchas polineuropatías inherentes no lo están (11). Podemos concluir, hasta el momento, que el proceso subyacente en el paciente es probablemente una polirradiculoneuropatía desmielinizante crónica adquirida.
La presencia de altos niveles de proteínas en el líquido cefalorraquídeo (LCR) y los hallazgos neurofisiológicos identificados apoyan la localización anatómica y fisiopatología del proceso planteado. Las raíces nerviosas cruzan a través del canal espinal y están rodeadas por LCR, relación que contribuye a las anomalías de este último.
Etiologías metabólicas, inflamatorias, infecciosas y neoplásicas podrían estar asociadas con polirradiculoneuropatía desmielinizante crónica adquirida (12). Los periodos de remisión seguidos de exacerbación de síntomas y la disociación albúmina-citológica identificada en el LCR debería incrementar la sospecha de un proceso inflamatorio. En adición, la ausencia de pleocitosis en el LCR aleja la sospecha de procesos infecciosos y neoplásicos. De esta forma, aproximamos nuestro diagnóstico a una polirradiculoneuropatía desmielinizante inflamatoria crónica (PDIC).
Recordemos que PDIC es un síndrome con muchas causas. Además de la presentación clásica idiopático (13), PDIC podría estar asociado con enfermedad del tejido conectivo, gamapatía monoclonal de significado incierto y discrasia de células plasmáticas (macroglobulinemia, síndrome POEMS, enfermedad de Castleman) (14).
Considerando el diagnóstico diferencial final y el hallazgo anatomo-patológico descrito en la presentación del caso, nuestra hipótesis diagnóstica es una PDIC asociada a enfermedad de Castleman.
Dr. Francisco Bravo Puccio (Dermatopatología)
El paciente acudió al servicio por presentar una lesión específica, una placa eritematosa pigmentada a nivel esternal, además de otras anomalías, como la hiperpigmentación difusa, hipertricosis y uñas en vidrio de reloj.
Por ser significativa e inusual, se procedió a tomar una biopsia de la placa pre-esternal. La histología mostró una hiperplasia capilar intersticial que correspondía a una neoproliferación vascular. Estas últimas se pueden dividir en las que corresponden a neoplasias (como el angiosarcoma o el sarcoma de Kaposi) y las que corresponden a procesos reactivos, como por ejemplo la verruga peruana (en el contexto de enfermedad de Carrión) o el hemangioma glomeruloide (asociado a POEMS).
Cuando hay una lesión de piel que corresponde a un proceso vascular proliferativo, en el contexto de una neuropatía periférica e hiperpigmentación, un diagnóstico a plantear inmediatamente es POEMS. Cabe anotar sin embargo, que el hemangioma glomeruloide, la lesión vascular clásica asociada a POEMS, es clínicamente una pápula angiomatosa (15), de tamaño menor a un centímetro, y no la placa descrita en este paciente.
Sin embargo, existen reportes de pacientes con síndrome de POEMS que presentaron desde el punto de vista semiológico, una lesión similar sino idéntica a la del paciente (16,17). En esos reportes, esta placa pre-esternal ha sido asociada a un plasmocitoma subyacente de localización esternal, el cual, para diagnóstico, hubiera requerido estudios de imágenes muy precisos, como son la tomografía, resonancia magnética o PET scan. Los otros hallazgos en el paciente, si bien menos específicos, son los mas frecuentemente asociados a S. POEMS, incluyendo hipertricosis, hiperpigmentación difusa y últimamente la lipoatrofia (18).
En conclusión, desde el punto de vista dermatológico, todas las lesiones cutáneas del paciente corresponden semiológicamente a lo que se puede encontrar en el síndrome de POEMS y variantes.
Dr. Cesar Ramos Aguilar (Dermatología)
Tres meses después del inicio de su enfermedad el paciente presenta varias alteraciones a nivel de la piel: lipoatrofia facial, y atrofia de interóseos, hiperpigmentación generalizada de la piel así como en las líneas de las manos, pigmentación de mucosas sin pigmentación gingival, piel indurada a la palpación, esclerodermiforme a predominio acral e hipertricosis que el nota con preocupación. Presenta uñas en vidrio de reloj (al oponer las uñas normales éstas forman una imagen en forma de rombo o ventana de Schamroth, él no tenía esa formación romboidal) y en faneras, ausencia de lúnula o uñas de Terry (síndrome de uñas blancas).
La lesión en el tórax anterior, podemos describirla como una lesión tipo mancha o parche plana, eritematosa purpúrea, con aumento de calor, asintomática y con vasos superficiales tipo telangiectasias en el borde de la lesión; por estos hallazgos se sospecha de una proliferación de origen vascular siendo el diagnóstico diferencial eritema crónico migrans (enfermadad por Borrelia), Mucinosis Reticular Eritematosa REM (forma de lupus cutáneo), eritema pigmentado fijo (toxicodermia), linfoma cutáneo (poiquilodermia atrófica vascular, manifestación de micosis fungoide). Con la evolución la piel se torna ictiosiforme, dato clínico importante.
¿Estos cambios cutáneos y la proliferación vascular se presentan en el síndrome de POEMS? Los reportes han sugerido una relación entre las lesiones cutáneas y los niveles altos de VEGF, secretados por esta discrasia de células plasmáticas (19). Las lesiones cutáneas asociadas a POEMS se presentan en 50 a 90% de los pacientes. Estas son: la hiperpigmentación difusa (93%), hipertricosis (78%), cambios esclerodermiformes (85%), acropaquia y acrocianosis, uñas de Terry y proliferaciones vasculares tipo parches hasta angiomas (26-44%) (2,19).
El hemangioma glomeruloide fue descrito por primera vez en 1990 por Chan y col. Clínicamente se caracteriza por múltiples pápulas o nódulos rojos purpúreos en región pectoral y extremidades superiores. Siendo considerado un marcador cutáneo del síndrome de POEMS, no es exclusivo, encontrándose en pacientes con Enfermedad de Castleman, Crioglobulinemia y linfoma primario cutáneo.
Otra lesión vascular es la descrita tipo mancha o parche eritemato-purpúrico en tórax y extremidades teniendo un patrón de hemangioma microvenular, siendo posible que el plasmocitoma esté por debajo de esta lesión (AESOP: adenopathy and extensive skin patch overlying a plasmacytoma) y secrete factores angiogénicos que difundan a nivel local, explicando por qué la lesión se encuentra por encima del plasmocitoma. Este tipo de lesión es la que presentaba nuestro paciente, sin embargo no se encontró plasmocitoma o lesión osteoesclérotica en el esternón, mediante las radiografías simples. Tener en cuenta que la lesión dérmica precede al compromiso ganglionar dato importante para sospechar esta entidad (2,16,17).
La lipoatrofia facial se informó en pocos casos. El VEGF estaría involucrado en la regulación de la adipogénesis con incremento de IL-6 secretada por las células plasmáticas que participan en la lipólisis, siendo un efecto directo tóxico sobre el tejido adiposo (20).
La acropaquia también se debe al incremento VEGF y al factor de crecimiento derivado de las plaquetas provocando edema y por lo tanto la uña de Terry (2,20).
El Herpes virus 8 no se relaciona con la enfermedad de Castleman ni a las lesiones vasculares dérmicas asociadas al síndrome POEMS, en éste intervienen otros factores tales como la IL-6, que juega un papel en el desarrollo de los hemangiomas (21,22).
Como dato semiológico, en la intoxicación por plomo se aprecia el ribete gingival de Burton, signo que el paciente no lo presenta por lo que se aleja la posibilidad de intoxicación por plomo.
En conclusión, las manifestaciones cutáneas de este paciente son características del síndrome POEMS y es el principal diagnóstico a documentar con los otros criterios.
Dr. Diego Venegas Ojeda (Oncología)
Vamos a realizar dos apreciaciones: la primera respecto al diagnóstico y luego, al tratamiento. En cuanto al diagnóstico actualmente consideramos al Síndrome POEMS, como un síndrome paraneoplásico de una discrasia de células plasmáticas, que incluyen: Mieloma múltiple, plasmocitoma solitario óseo, plasmocitoma extramedular y gammapatía monoclonal de significado incierto (1,2).
La característica más importante en POEMS es la presencia de polirradiculoneuropatía inflamatoria crónica desmielinizante y el pico monoclonal que suele ser no tan elevado como lo vemos en Mieloma múltiple (2). Otras características ya han sido comentadas por las especialidades que me precedieron: Policitemia, endocrinopatías, incluso hipotiroidismo, lesiones cutáneas, lesiones osteoescleróticas, incremento del VEGF y la presencia de Enfermedad de Castleman que es una hiperplasia angiofolicular ganglionar de distribución centrípeta (23).
Este paciente tiene criterios para POEMS lamentablemente por cuestiones económicas, no se le ha podido hacer dosaje de inmunoglobulinas que ayudaría; sin embargo, en la inmunofijación en suero hay policlonalidad; por lo que el segundo criterio más importante para el diagnóstico no se cumple. Pero si revisamos la literatura hasta 25% de casos puede no encontrarse pico monoclonal, entonces también podría tratarse de esta forma de presentación (1).
En la evaluación de la medula ósea se observó 16% de células plasmáticas de aspecto anormal con nucléolos prominentes y eso implica que estamos frente a una discrasia de células plasmáticas
La enfermedad de Castleman es un desorden linfoproliferativo no clonal que puede afectar uno o más grupos ganglionares y donde la IL6 es el principal responsable en su fisiopatología. El virus herpes 8 codifica una citoquina homóloga a IL6, es por ello su presencia es casi una norma cuando tenemos un paciente con HIV y enfermedad de Castleman y por el contrario casi no lo encontramos en pacientes no HIV, como nuestro caso. Sin embargo, otros virus como Epstein bar y citomegalovirus también han sido asociados (24).
Se han descrito dos subtipos histopatológicos y clínicos el hialino vascular, donde predomina la expresión VEGF y EGFR y el subtipo células plasmáticas que usualmente acompaña al Síndrome POEMS (25,26).
En la Enfermedad de Castleman, la IL6 es la responsable del incremento de citoquinas proinflamatorias y respuesta TH2, que llevan al sujeto a la caquexia, e incrementa la expresión de VEGF. Se ha descrito un patrón de lesiones en piel violáceos en pacientes HIV (-) asiáticos y planteándose la hipótesis del polimorfismo genético (27).
POEMS se asocia en 10-30% a Enfermedad de Castleman; pero cabe destacar que los síntomas neurológicos son más severos en pacientes con POEMS que Enfermedad de Castleman (2).
Por lo expuesto, el diagnóstico más compatible con lo presentado por nuestro paciente es Síndrome POEMS secundario a una discrasia de células plasmáticas con enfermedad de Castleman no como diagnóstico principal; si no, como parte del cortejo clínico de POEMS.
En cuanto a la terapia actual revisemos lo que se reporta para Enfermedad de Castleman y POEMS. Se ha visto eficacia parcial de Rituximab en la enfermedad de Castleman variante hialino vascular, pero no para la de células plasmáticas, ya que el primero tiene una mayor expresión CD20 que la segunda (28). La quimioterapia convencional para linfoma logra respuestas del 50% en la Enfermedad de Castleman (26).
Si vemos terapias dirigidas a IL6 y otras citoquinas mediadoras tenemos: Suramin, un polisulfato de naftilurea usado para filariosis y tripanosoma, que bloquea el receptor de IL6; clínicamente no ha mostrado ser muy eficaz (26). Anakinra, antagonista del receptor de IL1, usado en Enfermedad de Still, bloquea el receptor de IL1 en el paso a NF-kappa B evitando la transcripción de citoquinas proinflamatorias como IL6; se ha visto tener actividad en enfermedad de Castleman diseminada en niños (28). Tocilizumab, es un anticuerpo monoclonal contra el receptor de IL6, en fase II desarrollado en Japón, ha demostrado rápida normalización de síntomas y de la linfadenopatía; sin embargo, al suspender la droga se observa exacerbación de la enfermedad (29). Siltuximab, es una anticuerpo quimérico que neutraliza la IL6 y que ha sido probado en fase I, logrando beneficio clínico en 78% de pacientes y respuesta radiológica en 52%; además de una respuesta dosis dependiente. El efecto adverso observado con estos anticuerpos ha sido las dislipidemias (26,30).
La terapia más recomendada es la quimioterapia en dosis altas, seguida de trasplante autólogo ya que logra una mejoría significativa del cuadro neurológico, respecto a las terapias anti-IL6, ya descritas (26).
En cuanto al Síndrome POEMS, se tiene en cuenta el principio para el manejo de los síndromes paraneoplásicos; es decir, lograr reducir los síntomas cuando se controla la enfermedad de fondo; en este caso, la discrasia de células plasmáticas.
En POEMS, los esteroides producen mejoría de los síntomas, pero su efecto es poco durable.
Con el uso de agentes alquilantes como ciclofosfamida, clorambucil o melfalán se ha logrado respuestas importantes y medianamente duraderas (10-19 meses). Li en China, usando melfalán en dosis de 10 mg/m2 y dexametasona 40 mg/día, los días 1 a 4 cada 28 días, reportó 81% de respuestas hematológicas en 31 pacientes y en todos logró respuesta neurológica y de niveles de VEGF al año de tratamiento (2,31).
Otros reportes también muestran respuestas a la quimioterapia a dosis altas con soporte de células madres; sin embargo, los criterios de inclusión no han sido homogéneos lo que impide generalizar los resultados (32,33, 34).
Nuevas drogas como bevacizumab, anticuerpo monoclonal contra el VEGF (factor de crecimiento del endotelio vascular) han sido probadas con respuestas moderadas; sin embargo, anasarca, ascitis, infecciones y falla multiorgánica han sido reportadas inicialmente (1).
El uso de Bortezomib, un inhibidor de proteosomas, usado como terapia estándar en mieloma múltiple, ha reportado mejoras del componente M, niveles de VEGF, polineuropatía y endocrinopatías en POEMS, por lo que se está realizando combinaciones con otros agentes para optimizar la respuesta (35,36).
El uso de talidomida y lenalidomida, que actúan reduciendo los niveles de IL6 y VEGF, ha mostrado respuestas moderadas; sin embargo, la neurotoxicidad de estas drogas debe ser evaluada de manera paralela (37-40).
Nuestro paciente empezó tratamiento con Dexametasona en dosis altas, mostrando mejora en la fuerza muscular pero los efectos secundarios nos obligan a buscar combinaciones con otros agentes alquilantes como melfalán o ciclofosfamida, este último disponible en el mercado, además de considerar la terapia con talidomida.
Editores
Es interesante la observación de dermatología del POEMS variante AESOP (adenopathy and extensive skin patch overlying a plasmacytoma), pues hace poco tuvimos en el servicio un paciente con diagnóstico de POEMS y una lesión tipo parche extensa en la región pectoral - muy parecida a la de nuestro paciente,- con un mieloma osteoesclerótico (MOS) en el esternón, debajo de la lesión dérmica. Este MOS fue demostrado por TEM de tórax y en ese caso si se tuvo pico monoclonal en la IF en sangre.
Evolución
En nuestro caso el paciente inició tratamiento con dexametasona 40 mg/día por 4 días semanal por 3 semanas cada 4 semanas, dos ciclos y luego continuó con ciclofosfamida 800 mg/m2 día 1, dexametasona 20 mg día 1- 4 cada 21 días por dos ciclos, presentando mejoría parcial. Luego se añadió talidomida 100 mg/día. Después de 4 meses de tratamiento el paciente ha logrado remisión completa de las lesiones en piel, la hemoglobina se encuentra en 16 g/dl, ha dejado la silla de ruedas y se desplaza apoyándose con muletas y en trayectos cortos de desplaza sin apoyo. Cabe resaltar que ha recibido concomitantemente tratamiento conjunto en Hematología, Endocrinología, Medicina Física y Rehabilitación; además, recibe metformina, levotiroxina y gemfibrozilo.
Diagnóstico final
Síndrome de POEMS
Enfermedad de Castleman Variante POEMS?
REFERENCIAS BIBLIOGRÁFICAS
1.Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003; 101(7):2496. [ Links ]
2.Dispenzieri A. POEMS syndrome: Update on diagnosis, risk stratification, and management. Am J Hematol. 2011; 86:592-601. [ Links ]
3.Gherardi RK, Bélec L, Soubrier M, et al. Overproduction of proinflammatory cytokines imbalanced by their antagonists in POEMS syndrome Blood. 1996; 87 (4):1458. [ Links ]
4.Watanabe O, Maruyama I, Arimura K, et al. Overproduction of vascular endothelial growth factor/vascular permeability factor is causative in Crow-Fukase (POEMS) syndrome. Muscle Nerve. 1998; 21 (11):1390. [ Links ]
5.Endo I, Mitsui T, Nishino M, Oshima Y, Matsumoto T. Diurnal fluctuation of edema synchronized with plasma VEGF concentration in a patient with POEMS syndrome. Intern Med. 2002; 41(12):1196. [ Links ]
6.Katzmann J, Kyle R, Benson J, et al. Screening Panels for Detection of Monoclonal Gammopathies. Clinical Chemistry. 2009; 55(8): 1517-1522. [ Links ]
7.Rajkumar SV, Dispenzieri A. Multiple myeloma and related disorders. In: Abeloff MD, Armitage JO, Niederhuber JE, Kastan MB, McKenna WG, eds. Abeloffs Clinical Oncology. 4th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2008. p. 2323-51. [ Links ]
8.Yoshizaki K, Matsuda T, Nishimoto N, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castlemans disease. Blood. 1989; 74 (4):1360. [ Links ]
9.Ropper AH, Raje NS, Lawrimore TM, Camelo-Piragua S, Sohani AR. Case records of the Massachusetts General Hospital. Case 7-2010. A 49-year-old man with peripheral neuropathy and ascites. N Engl J Med. 2010; 362 (10): 929-40. [ Links ]
10.Alport AR, Sander HW. Clinical approach to peripheral neuropathy: Anatomic localization and diagnostic testing. Continuum Lifelong Learning Neurol. 2012; 18(1):13-38. [ Links ]
11.Burns TM, Mauermann ML. The evaluation of polyneuropathies.Neurology. Clinical Practice 2011; 76 (S2):S6–S13. [ Links ]
12.Rubin DI. Acute and chronic polyradiculopathies. Continuum (Minneapolis Minn). 2011; 17(4):831–854. [ Links ]
13.Koller H, Kieseier BC, Jander S, Hartung HP. Chronic Inflammatory Demyelinating Polyneuropathy. N Engl J Med. 2005; 352:1343-56. [ Links ]
14.Katirji B, Koontz D. Disorders of peripheral nerves. In: Bradley WG, Daroff RB, Fenichel GM, Jankovic J, eds. Bradley: Neurology in Clinical Practice. Philadelphia: Elsevier/Saunders; 2012. p. 1915-2015. [ Links ]
15.Tsai CY, Lai CH, Chan HL, Kuo T. Glomeruloid hemangioma-a specific cutaneous marker of POEMS syndrome. Int J Dermatol. 2001; 40(6):403-6. [ Links ]
16.Lipsker D, Rondeau M, Massard G, Grosshans E. The AESOP (adenopathy and extensive skin patch overlying a plasmacytoma) syndrome: report of 4 cases of a new syndrome revealing POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes) syndrome at a curable stage. Medicine (Baltimore). 2003; 82(1):51-9. [ Links ]
17.Ganzetti G, Campanati A, Goteri G, Simonetti O, Offidani A.. Hemangioma in POEMS syndrome: an atypical clinical and histological presentation. J Eur Acad Dermatol Venereol. 2012; 26(7):927-8. [ Links ]
18.Barete S, Mouawad R, Choquet S, et al. Skin manifestations and vascular endothelial growth factor levels in POEMS syndrome: impact of autologous hematopoietic stem cell transplantation. Arch Dermatol. 2010; 146(6):615-23. [ Links ]
19.González-Guerra E, Haro MR, Fariña MC, Martín L, Manzarbeitia L, Requena L. Glomeruloid haemangioma is not always associated with POEMS syndrome. Clin Exp Dermatol. 2009; 34 (7):800-3. [ Links ]
20.Barete S, Mouawad R, Choquet S, et al. Skin manifestations and vascular endothelial growth factor levels in POEMS Syndrome impact of autologous hematopoietic stemcell transplantation. Arch Dermatol. 2010; 146(6):615-623 [ Links ]
21.Papo T, Soubrier M, Marcelin AG, et al. Human herpesvirus 8 infection, castlemans disease and POEMS syndrome. Br J Haematol. 1999; 104(4):932-3. [ Links ]
22.Obermoser G, Larcher C, Sheldon JA, Sepp N, Zelger B. Absence of human herpesvirus 8 in glomeruloid haemangiomas associated with POEMS syndrome and Castlemans disease. Br J Dermatol. 2003; 148(6):1276-8. [ Links ]
23.Cronin DM, Warnke RA. Castleman disease: an update on classification and the spectrum of associated lesions. Adv Anat Pathol. 2009; 16:236-246. [ Links ]
24.Naresh KN, Trivedi P, Horncastle D, Bower M. CD20 expression in the HHV-8-infected lymphoid cells in multicentric Castleman disease. Histopathology. 2009; 55:358-359. [ Links ]
25.Imai N, Taguchi J, Yagi N, et al. Relapse of polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes (POEMS) syndrome without increased level of vascular endothelial growth factor following successful autologous peripheral blood stem cell transplantation. Neuromuscul Disord. 2009; 19:363-365. [ Links ]
26.Van Rhe F, Stone K, Szmania S, Barlogie B, Singht Z. Castleman disease in the 21st century: an update on diagnosis, assessment and therapy. Clinical Advances in Hematology & Oncology. 2010; 8(7): 486-498. [ Links ]
27.Ocio EM, Sanchez-Guijo FM, Diez-Campelo M, et al. Efficacy of rituximab in an aggressive form of multicentric Castleman disease associated with immune phenomena. Am J Hematol. 2005; 78:302-305. [ Links ]
28.Galeotti C, Tran TA, Franchi-Abella S, Fabre M, Pariente D, Kone-Paut I. IL-1RA agonist (anakinra) in the treatment of multifocal castleman disease: case report. J Pediatr Hematol Oncol. 2008; 30:920-924. [ Links ]
29.Nishimoto N, Honda O, Sumikawa H, Johkoh T, Aozasa K, Kanakura Y. A Long-Term (5-Year) sustained efficacy of Tocilizumab for Multicentric Castlemans disease and the effect on pulmonary complications. Blood. 2007; 110(11): 646. [ Links ]
30.Nishimoto N, Kanakura Y, Aozasa K, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood.2005; 106:2627-2632 [ Links ]
31.Li J, Zhang W, Li J, et al. Combination of melphalan and dexamethasone for patients with newly diagnosed POEMS syndrome. Blood. 2011; 117(24):6445-6449. [ Links ]
32.Dispenzieri A, Moreno-Aspitia A, Suarez GA, et al. Peripheral blood stem cell transplantation in 16 patients with POEMS syndrome, and a review of the literature. Blood. 2004; 104:3400-3407. [ Links ]
33.Imai N, Taguchi J, Yagi N, et al. Relapse of polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes (POEMS) syndrome without increased level of vascular endothelial growth factor following successful autologous peripheral blood stem cell transplantation. Neuromuscul Disord. 2009; 19:363–365. [ Links ]
34.Kuwabara S, Misawa S, Kanai K, et al. Neurologic improvement after peripheral blood stem cell transplantation in POEMS syndrome. Neurology. 2008; 71:1691-1695. [ Links ]
35.Kaygusuz I, Tezcan H, Cetiner M, et al. Bortezomib: A new therapeutic option for POEMS syndrome. Eur J Haematol. 2010; 84:175–177. [ Links ]
36.Ohguchi H, Ohba R, Onishi Y, et al. Successful treatment with bortezomib and thalidomide for POEMS syndrome. Ann Hematol. 2011; 90:1113–4. [ Links ]
37.Jaccard A, Abraham J, Recher C, et al. Lenalidomide therapy in nine patients with POEMS syndrome. ASH Ann Meeting Abstr. 2009; 114:3872. [ Links ]
38.Kim SY, Lee SA, Ryoo HM, et al. Thalidomide for POEMS syndrome. Ann Hematol 2006; 85:545-546. [ Links ]
39.Kuwabara S, Misawa S, Kanai K, et al. Thalidomide reduces serum VEGF levels and improves peripheral neuropathy in POEMS syndrome. J Neurol Neurosurg Psychiatry. 2008; 79:1255-1257. [ Links ]
40.Sinisalo M, Hietaharju A, Sauranen J, et al. Thalidomide in POEMS syndrome: Case report. Am J Hematol. 2004; 76:66–68. [ Links ]
