










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Medica Herediana
Print version ISSN 1018-130X
Rev Med Hered vol.24 no.1 Lima Jan. 2013
Gran Ronda de Medicina Interna y Especialidades del Hospital Nacional Cayetano Heredia.
Caso clínico 01 - 2013. Mujer de 76 años con lesiones dérmicas, debilidad muscular, trastorno del sensorio y eosinofilia
Grand Round of Internal Medicine and Specialties at the Hospital Nacional Cayetano Heredia. Clinical case 01- 2013. A 76 year-old-woman with skin lesions, muscles weakness, altered mental status and blood eosinophilia
Editor de la sección: Dr. Sergio Vásquez Kunze Editores Asociados: Dr. Héctor Sosa Valle, Dr. Ray Ticse Aguirre, Dr. Leslie Soto Arquiñigo, Dra. Elena Zelaya Arteaga.
Sergio Vásquez1, Luis Vasquez2, Carolina Tokumura3, Armando Calvo4, Cristian León5.
1
Médico Asistente, Servicio de Medicina Interna. Dpto. Medicina. Hospital Nacional Cayetano Heredia. Lima, Perú.2
Médico Residente, Servicio de Medicina Interna. Dpto. Medicina. Hospital Nacional Cayetano Heredia. Lima, Perú.3
Médico Asistente, Servicio de Hematología-Oncología. Dpto. Medicina. Hospital Nacional Cayetano Heredia. Lima, Perú.4
Médico Jefe de Servicio de Reumatología. Dpto. Medicina. Hospital Nacional Cayetano Heredia. Lima, Perú.5
Médico Asistente, Servicio de Nefrología. Dpto. Medicina. Hospital Nacional Cayetano Heredia. Lima, Perú.
CASO CLÍNICO
Dr. Luis Herman Vásquez Silva (Residente de 3er Año de Medicina Interna)
Mujer de 76 años, natural y procedente de Cusco, jubilada, de ocupación anterior contadora, estado civil divorciada, quien 11 meses atrás notó la aparición de ampollas y placas eritematosas que evolucionaron a lesiones costrosas en hipogastrio pruriginosas, éstas se extendieron a miembros inferiores sin llegar a confluir. Luego aparecieron en brazos, cuello y pabellones auriculares. Acudió a un centro médico local donde le realizaron biopsia cutánea y le diagnosticaron dermatitis alérgica, recibiendo prednisona 60 mg diarios, corticoides tópicos y antihistamínicos, presentando mejoría parcial de las lesiones dérmicas y del prurito.
Ocho meses antes de la admisión ante la persistencia de los síntomas acudió a otro centro donde se le realizó una nueva biopsia de piel siendo en informe: "epidermis con ortoqueratosis, edema en dermis superficial, vasos capilares con paredes tumefactas e infiltrado inflamatorio perivascular superficial de linfocitos con ocasionales neutrófilos y abundantes eosinófilos que se extienden al intersticio", siendo el diagnóstico "hallazgos consistentes con urticaria-vasculitis". Le indicaron prednisona 20 mg diarios y continuaron el resto de la medicación. Las lesiones cutáneas persistieron con hiperpigmentación difusa a predominio de miembros inferiores.
Siete meses antes presentó pérdida de peso progresiva, y debilidad muscular y mialgia generalizada, presentando dificultad para incorporarse. Tuvo caída a nivel del piso que provocó fractura de los arcos costales del tercio medio de la parrilla costal derecha. La prednisona se descontinuó por indicación médica.
Seis meses antes presentó un episodio de pérdida de conciencia, siendo admitida en nuestro hospital. El examen físico reveló somnolencia, hipoactividad y no habían signos de focalización neurológica. En los exámenes de laboratorio se le encontró hiponatremia ( Tabla 1); recibió solución salina hipertónica, mejorando el sensorio completamente. Al alta se le indicó dieta hipoalergénica, suplementación de sal, restricción hídrica, antihistamínicos y control ambulatorio en consultorio externo.
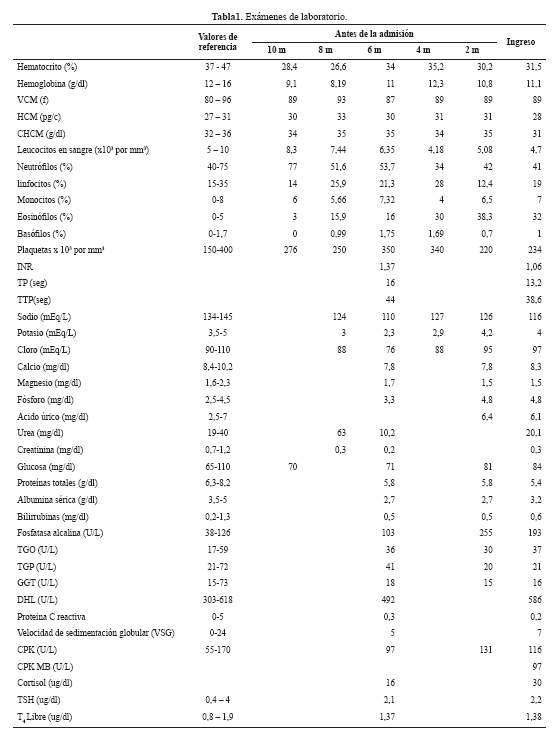
Cuatro meses antes, se agregó dificultad para levantar la cabeza, la debilidad empeoró requiriendo ayuda para la deambulación y subir escaleras, llegando a la postración y movilizándose solamente en silla de ruedas. Negaba parestesias o disestesias. En nuestro hospital se le realizó una nueva biopsia de piel siendo el informe dermatitis espongiótica con formación de vesículas espongioticas eosinófilas teniendo como diagnóstico diferencial eczema o pénfigo herpetiforme.
Dos meses antes del ingreso se realizaron exámenes adicionales (Tabla 1): ELISA para HTLV1 y VIH fueron no reactivos, el proteinograma electroforético sérico mostró hipoalbuminemia, hipogamaglobulinemia, incremento de la fracción alfa1 y leve incremento de la fracción alfa 2; IgA 155 mg/dl (VN 90 - 255), IgG 559 mg/dl (VN 700-1550); IgM 59 mg/dl (VN 40 - 260), ANA, ANCA y perfil ENA, Western Blot para Fasciola, IgM para Toxocara, parasicológico en heces seriado y Strongyloides (método de Baerman-Lumbreras) fueron negativos.
La radiografía y tomografía espiral multicorte (TEM) de tórax mostraron secuelas en lóbulos superiores y atelectasias mínimas en ambas bases. El TEM abdómino-pélvico solo evidenció engrosamiento concéntrico levemente irregular a nivel del cuerpo y antro de estómago. En este procedimiento presentó rash difuso a la administración del contraste, que se resolvió con una dosis de antihistamínicos y corticoides. Se le realizó endoscopía digestiva alta que mostró un divertículo esofágico y la colonoscopia la presencia de un pólipo colónico. La ecocardiografía transtorácica mostró hipertrofia septo basal no obstructiva, y esclerosis valvular mitral y aórtica de grado leve. La mamografía bilateral no tenía hallazgos significativos.
La resonancia magnética cerebral no evidenció proceso expansivo ni isquémico agudo. La electromiografía mostró velocidad de conducción nerviosa y potenciales de acción sensitiva normal, fibrilaciones y ondas positivas en regular cantidad, unidades motores polifásicas de amplitud y duración disminuida de aspecto miopático con patrón de interferencia completo, compatible con miopatía con denervación sugerente de miopatía inflamatoria.
Ante el empeoramiento de la debilidad, la persistencia de las lesiones cutáneas y el hallazgo nuevamente de hiponatremia en 116 mEq/L, se decidió su hospitalización.
En las funciones biológicas destacaba la hiporexia y la pérdida de 8 kg en los últimos 3 meses.
Tenía el antecedente de haber presentado dos episodios de hiponatremia sintomática hace 15 y 18 años, que no habían sido estudiados. Había sido diagnosticada hace 20 años de arritmia sinusal con ectopia supraventricular y ventricular medicada con atenolol, y cambiada a propanolol tres meses antes del ingreso y descontinuado dos semanas antes. Había sido hospitalizada por mordedura de serpiente hace 50 años.
Tuvo un solo embarazo con parto vaginal sin complicaciones y su última regla fue a los 52 años. La paciente negaba historia de hipertensión arterial, diabetes, fiebre de Malta, hepatitis, tuberculosis, contactos tuberculosos, no había recibió transfusión sanguínea previa. Se le había realizado quistectomía y oforectomía unilateral hace 40 años. Refería ser alérgica a la penicilina y a las sulfas. Su hermana falleció de cáncer de colon a los 75 años y su madre tuvo psoriasis. Estaba viviendo en Lima desde hace 9 meses debido al estudio de su enfermedad actual. Vivía con su hija quien la asistía.
La revisión anamnésica por aparatos y sistemas reveló disminución de la audición y sensación de rigidez en extremidades.
Al examen físico tenía presión arterial 100/60 mm Hg, frecuencia cardíaca 78 por minuto, frecuencia respiratoria 16 por minuto, estaba afebril, peso 34 kg e IMC 13. Lucía crónica y severamente enferma, en regular estado general, en mal estado de nutrición. La piel tenía zonas con descamación y lesiones costrosas hiperpigmentadas difusas, algunas lesiones vesiculares de fondo mielicérico en piernas; el tejido celular subcutáneo estaba muy disminuido y tenía atrofia muscular proximal y distal. Presentaba ganglios de 0,5 a 1 cm de diámetro en región inguinal, móviles no dolorosas. El examen de cabeza no era significativo y en el cuello no se palpaba la tiroides. No se encontró anormalidad cardiovascular salvo extrasístoles esporádicas, el aparato respiratorio era normal, en el abdomen no había visceromegalia y el examen genitourinario era normal. Al examen neurológico presentaba trofismo muscular muy disminuido incluyendo interóseos, pero con tono conservado, la fuerza muscular estaba disminuida (4-/5) tanto distal como proximal, había debilidad de la musculatura cervical que producía caída de la cabeza hacia adelante y presentaba imposibilidad para mantenerse en pie. No había fasciculaciones ni mioclonía y los reflejos estaban conservados al igual que la sensibilidad. El resto del examen era normal.
Los exámenes auxiliares realizados en la admisión se muestran en la tabla 1. Además, el examen de orina mostró pH 7, densidad 1010 y sedimento normal; proteinuria en orina de 24 horas 268 mg, osmolalidad sérica 244 mOsm/Kg agua, osmolalidad urinaria 191 mOsm/Kg agua, Na orina 74,7 mEq/L, K orina 30,4 mEq/L, ADH 10,88 pg/mL (VN: 0.10-8), vitamina B12 944 pg/mL (VN: 160-800), ácido fólico 4,29 ng/mL (VN: 3-17), vitamina D 8,6 nmol/L (VN: >30), ferritina sérica 360 ng/mL (VN: 14-233). Un nuevo examen de velocidad de conducción nerviosa con pruebas de electro estimulación para despistaje de miastenia gravis y síndrome miasténico de Lambert-Eaton, resultó negativo. El dosaje de anticuerpo contra el receptor de acetilcolina resultó negativo.
DISCUSIÓN
La discusión se centró sobre el diagnóstico etiológico y sobre los procedimientos claves o exámenes auxiliares para llegar al diagnóstico. Se invitó a participar a los médicos de los servicios de medicina interna, reumatología, hematología y nefrología.
Dr. Sergio Vásquez Kunze. (Medicina Interna)
Esta paciente presenta una historia compleja de múltiples síntomas, signos y alteraciones en los exámenes de laboratorio importantes. Comenzaría por reconocer los principales problemas, discutirlos separadamente y analizar si pueden formar parte de una misma enfermedad. Los cuatro problemas cardinales que presenta la paciente son: miopatía acompañada de caquexia, lesiones dérmicas, hipereosinofilia e hiponatremia crónica.
Miopatía
La miopatía de esta paciente tiene un componente proximal evidenciado por la debilidad de la musculatura cervical que lleva a la caída de la cabeza hacia delante (head drop) y por las caídas e imposibilidad de levantarse; pero también tiene gran atrofia de la musculatura distal, como los interóseos. La electromiografía con ondas de fibrilación y actividad polifásica de las unidades motoras son altamente sugestivas de miopatía inflamatoria. Si bien la CPK estaba normal esto puede observarse en un pequeño porcentaje de éstas miopatías como pudiera ser el caso de la paciente (1).
Dos son las miopatías inflamatorias a comentar: Miositis de cuerpos de inclusión (MCI) y polimiositis (PM). Por la edad la MCI sería la mas frecuente, sin embargo, hay características que presenta la paciente en contra de esta posibilidad. La primera es que la MCI es una enfermedad crónica, de larga evolución y debuta con compromiso de la musculatura distal, aunque también puede debutar con caídas por debilidad en los cuadriceps. La segunda es que se asocia poco con compromiso sistémico, autoinmune o paraneoplásico, como parece tener esta paciente. Para PM tiene varias características a favor. El curso de meses es más típico de PM y la debilidad proximal característica. Tiene asociación frecuente con enfermedades sistémicas autoinmunes, de tejido conectivo y retrovirus. Su asociación con neoplasia es debatible en comparación con la dermatomiositis (DM), que está fuertemente asociada. Entre las enfermedades sistémicas con manifestación dermatológica que se asocian con PM destacan la dermatitis herpetiforme, psoriasis, acné fulminans, lupus y el síndrome hipereosinofílico (SHE) (2). La paciente tiene hipereosinofilia y lesiones dérmicas entonces, PM es un diagnóstico importante a considerar.
Aunque esta paciente estuvo recibiendo dosis altas de prednisona, la miopatía por corticoides por lo general no muestra el patrón en la EMG ni produce un cuadro tan severo como el que presenta la paciente.
Otro diagnóstico a considerar es miositis hipereosinofílica. La hipereosinofilia puede infiltrar diversos órganos no excluyéndose el músculo esquelético, aunque esta presentación es muy rara (2).
Por último, la gran caquexia con sarcopenia severa también es un diagnóstico diferencial y de descarte. Para el diagnóstico de estas patologías es indispensable la biopsia de músculo con las técnicas adecuadas.
Lesiones dermatológicas
Las lesiones dérmicas han sido difusas, eczematosas y al inicio bulosas con hiperpigmentación postinflamatoria. Una constante en las biopsias de piel ha sido la infiltración de eosinófilos. Esta infiltración es inespecífica y se puede ver en enfermedades bulosas, alérgicas, reacción a drogas, picaduras de insectos, mastocitosis sistémica y sindromes hipereosinofílicos (3-5). Como el paciente tiene hipereosinofilia y compromiso sistémico debe sospecharse ésta entidad como causa de la infiltración dérmica por eosinófilos.
Hipereosinofilia
El diagnóstico de eosinofilia se establece con una cuenta mayor de 500/mm3 y de hipereosinofilia cuando es mayor de 1500/mm3 en 2 ocasiones separadas (6). Con excepción del hemograma de hace 10 meses, en todos los demás la presenta. Es probable que en el primer hemograma no la haya presentado debido al uso de esteroides, que tiene un efecto eosinopénico notable.
La importancia de diferenciar una eosinofilia de una hipereosinofilia estriba que ésta última pone al paciente en mayor riesgo de diagnósticos etiológicos de tipo neoplásico, autoinmunes o sistémicos, y no solo alergias, eczemas o infecciones parasitarias típicamente helmintos ( Tabla 2). En la hipereosinofilia también hay un riesgo importante de infiltración de órganos blanco por eosinofilos y daño de éstos por sus proteínas tóxicas, como la catiónica y básica mayor. Cuando la hiperesonofilia se asocia a infiltración y daño de tejidos se denomina síndrome hipereosinofílico (SHE) (6). Este puede ser primario si es de origen clonal de precursores medulares de eosinófilos con mutaciones oncogénicas de receptores de la tirosin kinasa, como el platelet derived growth factor receptor A (PDGFRA), platelet derived growth factor receptor B (PDGFRB) y el fibroblast growth factor receptor 1 (FGFR1); o secundario, es decir reactivo, como en el SHE linfocítico, pero también lo pueden producir parásitos o linfomas (7).

Así, cuando estamos frente a un paciente con hipereosinofilia hay doble tarea, buscar la etiología y si hay daño en órgano blanco. Se debe hacer una historia cuidadosa que incluya procedencia (regiones tropicales asociadas a parasitosis), uso de fármacos, asma y alergias como los más resaltantes. El examen físico debe buscar signos de compromiso sistémico o que involucre infiltración de órgano blanco como piel, aparato gastrointestinal, corazón o pulmones, que son los más típicos. Luego el plan debe incluir repetir el hemograma, estudios parasitológicos en heces (especialmente pruebas de alta sensibilidad para Strongyloides) y pruebas que busquen daño en órgano blanco como pruebas de función hepática, renal y pulmonar, enzimas cardiacas, EKG e imágenes como ecocardiografía y TEM de tórax y abdomen. Las biopsias de tejidos y otros estudios (como serología para Fasciola hepática, Toxocara cannis, ANCA como marcador de vasculitis de Churg-Strauss, VIH) deben hacerse si están clínicamente indicados.
La paciente no tenía diarrea ni procedía de zonas de mayor riesgo para strongiloidiasis ni infecciones por helmintos y los exámenes parasitológicos y para HTLV-1, fueron negativos. No tenía factores de riesgo ni colestasis para sospechar fasciolasis, ni historia de riesgo de exposición a toxocara, y los estudios serológicos para estas entidades fueron negativos. No tenía compromiso pulmonar para pensar en vasculitis de Churg-Strauss, y el ANCA fue negativo. No había recibido drogas nuevas al inicio de su enfermedad que justifique una reacción adversa a fármacos y las biopsias de piel no eran diagnósticas de mastocitosis ni de pénfigo. Todos estos estudios resultaron negativos para considerar un SHE secundario. La infiltración de órganos era evidente en la piel y posiblemente cardiaco por engrosamiento del septum y la presencia de arritmias.
Faltaría descartar en la paciente, el SHE primario (clonal) y la variante linfocítica que es reactiva (Tabla 2). Estos necesitan pruebas de RT-PCR para mutaciones, citogenética y citometría de flujo (CF) para acercarnos al diagnóstico de las variantes mieloide (primaria) y linfoide (reactiva) del SHE. Entre las pruebas disponibles en nuestro medio tenemos: fusión FIP1L1-PDGFRA, mutación c-KIT D816V y traslocación ETV6 (TEL)-PDGFRB para diagnóstico de la forma mieloide, y CF con demostración CD3 negativo CD4 positivo para la variante linfoide. El estudio cromosómico debe evaluar si hay cromosoma filadelfia por la posibilidad de presentación de leucemia mieloide crónica con hipereosinofilia.
Hiponatremia
La hiponatremia es euvolémica, persistente y crónica, y tiene una osmolalidad urinaria mayor de 100 mOsm/kg de agua, con niveles de cortisol y de hormonas tiroideas normales, la cual hace el diagnóstico de síndrome de secreción inapropiada de hormona antidiurética (SIADH), probablemente paraneoplásico. Nótese la gran cronicidad de la hiponatremia que puede asociarse a neoplasias indolentes y de curso larvado como los síndromes hipereosinofílicos.
En conclusión, se debe hacer biopsia de músculo, estudios de mutaciones y CF. Dada la gran posibilidad de un SHE primario y que la paciente esta en constante deterioro estaría de acuerdo en iniciar corticoides a dosis altas. Los SHE primarios variante linfoide tienen una buena respuesta para controlar la eosinofilia y también sería de utilidad de ser una PM.
Dr. Armando Calvo Quiroz (Reumatología)
La debilidad muscular de la paciente es ocasionada por miopatía, lo cual se sustenta en la anamnesis, el examen físico y la electromiografía. La electromiografía fue sugerente de miopatía inflamatoria, mostrando unidades motoras polifásicas de amplitud y duración disminuida, fibrilaciones y ondas positivas en regular cantidad, con patrón de interferencia completo. No se evidenció elevación de enzimas musculares, lo cual puede suceder hasta en 10% de las miopatías inflamatorias o estar condicionado por una gran pérdida de masa muscular, la administración de corticoides, alcoholismo o hipertiroidismo.
Empleando criterios clínico-patológicos las miopatías inflamatorias se pueden clasificar en seis categorías: Dermatomiositis, polimiositis, síndromes de sobreposición con otras enfermedades del tejido conectivo, miositis asociadas a cáncer, miositis por cuerpos de inclusión y otras formas que incluyen variantes raras focales y difusas (8).
Como la paciente presenta manifestaciones dermatológicas, es posible plantear el diagnóstico de dermatomiositis; sin embargo, las lesiones morfológicamente no corresponden a las lesiones patognomónicas de esta enfermedad como el heliotropo o el gottron y tampoco a las específicas, además, las biopsias cutáneas muestran infiltración eosinofílica importante, lo que no es característico de la dermatomiositis.
La presencia de manifestaciones extramusculares como el cuadro dérmico, la hipereosinofilia, el estado consuntivo, el trastorno electrolítico y el compromiso de conciencia señalan la presencia de un cuadro sistémico, lo que aleja la posibilidad de polimiositis y miopatía por cuerpos de inclusión; ambos cuadros clínicos habitualmente están centrados en las manifestaciones musculares, aunque resulta atractivo el plantemiento de miopatía por cuerpos de inclusión por la edad de la paciente, el cuadro consuntivo y el gran componente distal en la debilidad muscular. La investigación de marcadores inmunológicos de autoinmunidad alejó la posibilidad de una sobreposición.
Por tanto la clínica y la pobre respuesta a la terapia corticoide hacen que el diagnóstico más probable sea el de una miositis asociada a cáncer, planteándose adicionar a los estudios ya realizados, la investigación de la hipereosinofilia y la realización de biopsia muscular.
Dra. Carolina Tokumura Tokumura (Hematología)
La paciente presenta recuentos de eosinófilos mayores a 1 500 leucocitos x mm3, cumpliéndose la definición de hipereosinofilia. Si ésta provoca daño de órgano blanco, es decir fibrosis endomiocardica, encefalopatía, trombosis o patología gastrointestinal, debe ser tratada independientemente de la causa por la severidad de las complicaciones observadas y porque en algunos casos provocan daño permanente. Con respeto a la etiología, primero se deben descartar las causas secundarias de hipereosinofilia; en la paciente los eosinófilos se incrementan 8 meses antes de la admisión y podría relacionarse a cambios en la medicación, por lo que debe descartarse una reacción adversa a drogas.
Si se tratara de una hipereosinofilia variedad linfocítica, podría responder a corticoides pues es mediada por citoquinas, lo que no pasaría con la variedad mieloide que se trata de una expansión clonal por la presencia de alteraciones cromosómicas. Se debe realizar estudios moleculares y citogenéticos en la medula ósea buscando genes de fusión con actividad tirosin kinasa y descartar presencia del cromosoma filadelfia.
Dr. Cristian León Rabanal (Nefrología)
Un detalle resaltante en la historia de esta paciente es definitivamente la hiponatremia. Como hallazgo aislado tal vez no brinde mayor información que la ya conocida en la literatura; distinto es el caso en el cual la hiponatremia es parte de una enfermedad con manifestaciones sistémicas marcadas.
En este caso tenemos varios indicios para sospechar de una hiponatremia crónica en una paciente adulta mayor; rápidamente el posible diagnóstico de SIHAD aparece como la posibilidad diagnóstica más oportuna y que es corroborada por la inapropiada osmolaridad urinaria en una paciente con hiposomolalidad sérica. Se describen hasta 4 tipos clasificados con las letras A B C y D diferenciándose en el patrón de secreción de la ADH. Por la edad y por el estado nutricional de la paciente nos inclinamos por el tipo B o reset osmostat, pero lo más importante es el problema que está detrás de la aparición de este trastorno.
El SIHAD es producido por múltiples trastornos clínicos. Los más frecuentes son los fármacos y las neoplasias. Dado los síntomas y signos de esta paciente podríamos sugerir una hiponatremia crónica posiblemente como una manifestación paraneoplásica de un tumor o neoplasia oculta y que debe ser evaluada en profundidad. Puede haber una serie de razones para que ocurran episodios de exacerbación, la mayoría de ellos relacionadas a la ingesta inadecuada de agua libre.
Llama la atención el fósforo sérico en límites normales. Siendo una paciente desnutrida este hallazgo nos sugiere una posible patología tubular distal con alteración del aclaramiento del fosfato, requiriendo una depuración de creatinina para determinar si existe compromiso renal y se podría relacionar también a liberación de fosforo intracelular de una patología neoplásica que también podría estar en relación al SIHAD (9-11).
EVOLUCIÓN
Se le realizó una biopsia muscular de deltoides encontrándose atrofia muscular moderada sin incremento de células inflamatorias, siendo compatible con denervación. Luego, una RMN de cintura pelviana solo evidenció marcada atrofia muscular sin alteraciones de su señal. Una nueva biopsia de musculo en cintura escapular volvió a revelar atrofia de fibras tipo II con denervación leve reciente, sin cambios de miositis o cuerpos de inclusión. El estudio de médula ósea no fue evaluable por falta de espículas. El informe de la biopsia de hueso indicó "ausencia de tejido hematopoyético con grupos de eosinófilos y algunos linfocitos". Se decidió iniciar Prednisona a 1 mg/kg/d.
Ante presencia de hipereosinofilia y habiéndose realizado estudios que descartaron que fuera de causa reactiva, se procedió con estudios de citogenética los cuales mostraron: mutación c-KIT D816V: no mutado, transcriptos de fusión FIP1L1-PDGFRA: negativo, traslocación ETV6 (TEL)-PDGFRB: positivo. Citometría de flujo: CD3: 657 cel/mm3, CD3/CD4: 460 cel/mm3, CD3/CD8: 160 cel/mm3, relación CD4/CD8: 2,87.
COMENTARIOS
Dr. Sergio Vásquez Kunze
Dado lo infrecuente del diagnóstico y de su demostración molecular quisiera resumir la clasificación de los SHE, sus características clínicas y tratamiento. Ya mencione que una forma de clasificarlo es primario, secundario e idiopático. Dentro de los primarios está la forma mieloproliferativa (SHE-M o también llamada neoplasia mieloproliferativa) y en los secundarios hay que es resaltar la variante linfocítica (SHE-L); hay también familiares, asociados y overlap, pero éstos no encajan en nuestro paciente.
Hay que recordar que hay 2 formas de que haya proliferación de los eosinófilos: reactivamente por mediación de la interleuquina 5 (IL-5), el GM-CSF e IL-3, todos éstos son secretados por linfocitos T activados (12,13); y mediante una clona per se (neoplasia) de eosinófilos, células mieloides o stem cell (14).
El SHE-L se produce por una clona de linfocitos T que produce IL-5 (15), que a su vez estimula la proliferación de eosinófilos (por eso se dice que es secundaria, no es una clona de eosinofilos neoplásica). En el SHE-L el cuadro clínico es crónico, principalmente infiltración dérmica, con rash y prurito, y edema (15). Se confunde a veces con reacciones alérgicas y urticaria crónica. Puede presentar infiltración de órganos pero es infrecuente. Típicamente se hace el diagnóstico por presentar en sangre periférica linfocitos T CD3 negativo y CD4 positivo. La respuesta a corticoides es muy buena, disminuyendo los eosinófilos en horas y las manifestaciones dérmicas y el edema. Luego se debe ir retirando los esteroides y pueden agregarse otras drogas como hidroxiurea o interferon alfa (16,17). Clínicamente esta paciente parecía tener esta variante por el compromiso dérmico y la buena respuesta a esteroides pero no tuvo el fenotipo característico en la CF.
El SHE-M se origina por una clona de precursores de eosinófilos con mutaciones oncogénicas de receptores de la tirosin kinasa como el platelet derived growth factor receptor A (PDGFRA) que es el más común, platelet derived growth factor receptor B (PDGFRB) y el fibroblast growth factor receptor 1 (FGFR1)(7,14). Esta forma típicamente puede infiltrar órganos u ocasionar necrosis y fibrosis de éstos como ya se mencionó. La frecuencia de afectación de órganos es: piel (37%), pulmonar (25%), gastrointestinal (14%) y cardiaca (5%) (16). Sin embargo, la afección cardiaca es la que causa mayor morbimortalidad, esta pasa por un estadio de necrosis endomiocárdica, trombosis intracardiaca y fibrosis (18). No es inusual que el paciente debute con un cardioembolismo cerebral, como hemos visto en nuestro hospital en otro paciente. El componente cardiaco esta más asociado a la mutación del PDGFRA que además es más agresiva que la PDGFRB.
La paciente presenta translocación ETV6 (TEL)-PDGFRB; como ya mencionamos es menos frecuente que la fusión FIP1L1-PDGFRA. Ambas fusiones activan los genes para PDGRF que codifican el receptor de tirosin kinasa. La actividad de éstas está asociada a enfermedad mieloproliferativa con hipereosinofilia. En una serie de 4 casos de SHE asociado a translocación ETV6 (TEL)-PDGFRB se mostró que ésta era una enfermedad muy crónica y en un paciente tenía 13 años de evolución. La mayoría de ellos tenía lesiones dérmicas y podía o no presentar esplenomegalia o citopenias (19).
Con referencia al tratamiento, el imatinib es una droga que inhibe la actividad de la kinasa del BCR-ABL, PDGRFA, PDGFRB y c-KIT. Hay mucha más experiencia y éxito en inhibir la actividad de BCR-ABL y PDGRFA que en la de PDGFRB (20-23). Sin embargo, en la serie de 4 casos con translocación del PDGRFB mencionada todos tuvieron buena respuesta a imatinib 400mg/d, cediendo la eosinofilia y las lesiones dérmicas, habiendo fracasado a terapias inespecíficas como hidroxiurea, pipobroman e interferon alfa (19). En conclusión la paciente tiene indicación para añadir imatinib a los corticoides.
Debo añadir que los SHE-M se deben diferenciar de la leucemia mieloide crónica (LMC) con eosinofilia mediante la búsqueda del cromosoma filadelfia, aunque la LMC frecuentemente presenta otras citopenias, hepatoesplenomegalia, vitamina B12 elevada, que no estaban presentes en nuestra paciente. También se debe diferenciar de la mastocitosis sistémica con eosinofilia, buscando la mutación del c-kit que se presenta en ésta última y que estaba ausente en nuestra paciente. Por último, la leucemia eosinofílica crónica se caracteriza por la presencia de blastos en menos del 20% en la médula ósea.
Dra. Carolina Tokumura Tokumura
Los estudios moleculares demostraron la presencia de una translocación ETV6 (TEL)-PDGFRB, gen de fusión poco frecuente, con actividad de tirosin kinsasa, asociado a síndromes mieloproliferativos, lo que explicaría la proliferación de eosinófilos y apoyaría el diagnostico de síndrome hipereosinofílico variante mieloproliferativa y probablemente sensible a imatinib con dosis de 100 a 400 mg día (24,25). La respuesta del imatinib se espera en 2 a 6 semanas. Lamentablemente no se obtuvo el estudio del cariotipo para descartar otras translocaciones (cromosoma filadelfia) y la citometria de flujo no demostró clonas anormales de estirpe linfoide. Llama la atención la buena respuesta a corticoides siendo una hipereosinofilia variante mieloide.
Dr. Armando Calvo Quiroz
Los hallazgos de la biopsia muscular descartan la posibilidad de miositis por cuerpos de inclusión, y no muestra la característica inflamación endomisial de la polimiositis o la inflamación perimisial de la dermatomiositis. Tampoco la resonancia magnética mostró inflamación muscular.
La investigación de la hipereosinofilia descartó que esta fuera reactiva y el estudio citogenético mostró la existencia de translocación ETV6 (TEL)-PDGFRB, lo cual indica que se trata de una hipereosinofilia asociada a malignidad.
Por ello planteamos que la debilidad muscular ocurra como un cuadro paraneoplásico en el cual como se observa en la biopsia ocurra denervación asociada a atrofia y no un cuadro característico de dermatomiositis o polimiositis.
El caso nos deja varias enseñanzas, la primera, que el enfoque del paciente por problemas es imprescindible cuando no identificamos de primera intención la entidad nosológica o la patogenia de la enfermedad que lo afecta, y que la interacción entre las diversas especialidades de la medicina interna es fundamental en este enfoque. También debemos resaltar la importancia de incorporar la biología molecular en el diagnóstico y tratamiento de los pacientes. Finalmente, la importancia de la participación activa del paciente y su familia en el proceso diagnóstico y terapéutico, debiendo en este caso agradecer al familiar de nuestro paciente por su paciencia y dedicación.
EVOLUCIÓN FINAL
Con la terapia con prednisona 1mg/kg/día, se evidenció caída progresiva del valor de los eosinófilos llegando a un valor de 0,5% después de 3 días de tratamiento. Luego se inició imatinib 400 mg al día. La paciente continuó con restricción hídrica y suplementación de sal. Además inició terapia física.
La paciente tuvo mejoría de la fuerza de extremidades y de región cervical pero sin llegar a deambular sola. Aumentó 5 kilos de peso y las lesiones dérmicas disminuyeron.
Se disminuyó progresivamente la dosis de prednisona hasta 15 mg al día e imatinib a 200 mg por día. Posteriormente la paciente volvió a perder peso y cursó con un desorden depresivo por lo cual recibe escitalopram. Actualmente la paciente se mantiene aún con debilidad muscular.
DIAGNÓSTICO FINAL
- Síndrome hipereosinofílico variante mieloproliferativa traslocación ETV6 (TEL) PDGFRB: positivo.
Caquexia y atrofia muscular paraneoplásica. ¿Miositis paraneoplásica?
Secreción Inapropiada de hormona antidiurética crónica asociada a malignidad.
REFERENCIAS BIBLIOGRÁFICAS
1. Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977; 56(4):255. [ Links ]
2. Layzer RB, Shearn MA. Eosinophilic polymyositis. Ann Neurol. 1977; 1: 65-71. [ Links ]
3. Uehara M, Izukura R, Sawai T. Blood eosinophilia in atopic dermatitis. Clin Exp Dermatol. 1990; 15: 264. [ Links ]
4. Leiferman KM. Eosinophils in atopic dermatitis. J Allergy Clin Immunol. 1994; 94:1310. [ Links ]
5. McEvoy MT, Peterson EA, Kobza-Black A, et al. Immunohistological comparison of granulated cell proteins in induced immediate urticarial dermographism and delayed pressure urticaria lesions. Br J Dermatol. 1995; 133:853. [ Links ]
6. Simon HU, Rothenberg ME, Bochner BS, et al. Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol. 2010; 126:45. [ Links ]
7. Swerdlow SH, Campo E, Harris NL, et al. World Health Organization classification of tumours of haematopoietic and lymphoid tissues. 4° Edition. Lyon: IARC Press; 2008. [ Links ]
8. Mastaglia FL, Phillips BA. Idiopathic inflammatory myopathies:epidemiology, classification,and diagnostic criteria. Rheum Dis Clin N Am. 2002; 28: 723–741. [ Links ]
9. David H. Ellison DH, Berl T. The Syndrome of Inappropriate Antidiuresis. N Engl J Med. 2007; 356: 2064-2072. [ Links ]
10. Adrogue HJ, Madias NE. Hyponatremia. N Engl J Med. 2000; 342:1581-1589. [ Links ]
11. Hawkins RC. La edad y el género como factores de riesgo de hiponatremia e hipernatremia. Clin Chim Acta. 2003; 337:169-172. [ Links ]
12. Gleich GJ. Mechanisms of eosinophil-associated inflammation. J Allergy Clin Immunol. 2000;105:651- 63. [ Links ]
13. Ackerman SJ, Bochner BS. Mechanisms of eosinophilia in the pathogenesis of hypereosinophilic disorders. Immunol Allergy Clin North Am. 2007; 27:357-75. [ Links ]
14. Bain BJ, Fletcher SH. Chronic eosinophilic leukemias and the myeloproliferative variant of the hypereosinophilic syndrome. Immunol Allergy Clin North Am. 2007; 27:377. [ Links ]
15. Roufosse F, Cogan E, Goldman M. Lymphocytic variant hypereosinophilic syndromes. Immunol Allergy Clin North Am. 2007; 27:389. [ Links ]
16. Ogbogu PU, Bochner BS, Butterfield JH, et al. Hypereosinophilic syndrome: a multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol. 2009; 124:1319. [ Links ]
17. Schandené L, Roufosse F, de Lavareille A, et al. Interferon alpha prevents spontaneous apoptosis of clonal Th2 cells associated with chronic hypereosinophilia. Blood. 2000; 96:4285. [ Links ]
18. Ogbogu PU, Rosing DR, Horne MK 3rd. Cardiovascular manifestations of hypereosinophilic syndromes. Immunol Allergy Clin North Am. 2007; 27:457. [ Links ]
19. Apperley J, Gardembas M, Melo JV, et al. Response to Imatinib Mesylate in Patients with Chronic Myeloproliferative Diseases with Rearrangements of the Platelet-Derived Growth Factor Receptor Beta. N Engl J Med. 2002; 347:481-487. [ Links ]
20. Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003; 348:1201. [ Links ]
21. Jovanovic JV, Score J, Waghorn K, et al. Low-dose imatinib mesylate leads to rapid induction of major molecular responses and achievement of complete molecular remission in FIP1L1-PDGFRA-positive chronic eosinophilic leukemia. Blood. 2007; 109:4635. [ Links ]
22. Cortes J, Ault P, Koller C, et al. Efficacy of imatinib mesylate in the treatment of idiopathic hypereosinophilic syndrome. Blood. 2003; 101:4714. [ Links ]
23. Pardanani A, Reeder T, Porrata LF, et al. Imatinib therapy for hypereosinophilic syndrome and other eosinophilic disorders. Blood 2003; 101:3391. [ Links ]
24. David M, Cross CP, Burgstaller S. Durable responses to imatinib in patients with PDGFRB fusion gene– positive and-ABL–negative chronic myeloproliferative disorders. Blood. 2007; 109:61- 64. [ Links ]
25. Peros-Golubicic T, Smojver-Jezek S. Hypereosinophilic Syndrome: Diagnosis and Treatment. Curr Opin Pulm Med. 2007; 13(5):422- 427. [ Links ]
Recibido: 28/01/13
Aceptado: 21/02/13
