










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Medica Herediana
On-line version ISSN 1729-214X
Rev Med Hered vol.24 no.4 Lima Oct./Dec. 2013
Estrategia de genotipado del gen FMR1: Método de diagnóstico alternativo para el Síndrome X Frágil y otras enfermedades por expansión de trinucleotidos
Genotyping strategy for the FMR1 gene: An alternative diagnostic method for the Fragile X syndrome and other trinucleotide expansion diseases
Saúl Lindo-Samanamud (1,2,a), Mario Cornejo-Olivas (1,3,b), Olimpio Ortega (1,a), Victoria Marca (1,c), Keren Espinoza-Huertas (1,2,d), Pilar Mazzetti (1,b)
(1) Servicio de Neurogenética, Instituto Nacional de Ciencias Neurológicas, Lima, Perú
(2) Escuela de Genética y Biotecnología y Facultad de Medicina, Universidad Nacional Mayor de San Marcos. Lima, Perú
(3) Northern Pacific Global Health Research Training Consortium, Bethesda, US.
(a) Biólogo
(b) Médico Neurólogo
(c )Magister en Bioquímica
(d) Bachiller en Genética y Biotecnología
RESUMEN
Objetivos: Diseñar una estrategia alternativa por PCR para el genotipado de secuencias ricas en citosinas, basada en modificación nucleotídica. Material y métodos: Se modificó el gen FMR1 nativo de ocho individuos clínicamente no afectados por el Síndrome X frágil, cambiando las citosinas por uracilos, empleando bisulfito de sodio. El ADN modificado fue purificado y cuantificado por espectrofotometría. Las estructuras alternativas y potenciales islas CpG que adopta el microsatélite inestable fueron simuladas con los programas MFOLD y CpGplot. Se generaron cebadores específicos que hibriden tanto con el microsatélite modificado (Primer T) y con una secuencia modificada de las islas CpG (Primer M), utilizando el programa MethPrimer. Finalmente, ambas secuencias fueron amplificadas por PCR y los amplicones fueron separados por electroforesis en gel de poliacrilamida (PAGE por sus siglas en inglés) al 6% y visualizados con tinción de nitrato de plata. Resultados: La modificación del ADN fue evidenciada por espectrofotometría al uracilo. Las estructuras observadas en la simulación fueron las horquillas encontrándose dos potenciales islas CpG. La amplificación con los cebadores T, confirmó el diseño in silico desarrollado para abordar la estructura en horquillas. La amplificación con los cebadores M permitió detectar metilación de la primera isla CpG del gen FMR1.Conclusión: Se propone un diseño alternativo para amplificación de secuencias de microsatélite que contengan citosinas metiladas y no metiladas. Se requieren estudios posteriores con muestras de ADN que contengan microsatélites muy expandidos para validar su aplicación para diagnóstico molecular.
PALABRAS CLAVE: Citosinas metilación, repetición de microsatélite, uracilo. (Fuente: DeCS BIREME)
SUMMARY
Objectives: To design an alternative strategy for genotyping cytosine-rich sequences using PCR and nucleotide modification. Methods: The FMR1 gene wild type was modified in the DNA obtained from eight individuals clinically unaffected for Fragile X Syndrome; cytosines were replaced by uracils using sodium bisulfite. Modified DNA was purified and quantified by spectrophotometry. Alternative structures and potential CpG islands of the unstable microsatellite were simulated using MFOLD and CpGplot tools. Specific primers were generated to hybridize with both the modified microsatellite (Primer G) and a modified sequence of CpG islands (Primer M) using the MethPrimer software. Finally, both sequences were amplified by PCR and the amplicons were separated by electrophoresis in silver-stained PAGE 6% gels. Results: The DNA modification was evidenced by spectrophotometry to uracil. We found two potential CpG islands. The amplification with T primers confirmed the "in silico" design developed to engage hairpin structures. The amplification with M primers detected methylation of the first CpG island in the FMR1 gene. Conclusion: We propose an alternative design for amplifying microsatellite sequences that contain methylated and unmethylated cytosine bases. Further studies are required with DNA samples containing expanded microsatellites to validate its molecular diagnostic application.
KEYWORDS: Cytosine, methylation, microsatellites repeats, uracil. (Source: MeSH NLM).
INTRODUCCIÓN
Existen varias enfermedades neurodegenerativas asociadas a tripletes repetidos, estas secuencias se convierten en microsatélites inestables (MI) cuando sus unidades repetitivas (UR) superan el rango normal (1). Estas secuencias tienden a adoptar múltiples conformaciones estructurales del ADN, alternativas a la convencional, que estarían relacionadas con la inestabilidad del microsatélite (mutación dinámica) y con el proceso de metilación del mismo (2).
El diagnóstico genético molecular de estos desórdenes se basa en la cuantificación de las UR de los MI en el gen responsable, utilizando metodologías como la reacción de cadena de la polimerasa (PCR), secuenciamiento y Southern blot, todas ellas con limitaciones para la cuantificación. La denaturación y amplificación de estos MI por PCR se vuelve ineficiente debido al gran número de UR y a la formación de estructuras alternativas (3). La metodología de Southern blot está considerada el estándar de oro para la cuantificación de UR; sin embargo, esta metodología es compleja, costosa y requiere manejo de sondas radioactivas (4).
En 1970, dos experimentos simultáneos que evaluaban la capacidad mutagénica del bisulfito de sodio en ADN de fago λ y E. coli sugieren el uso potencial de esta molécula para la modificación de los ácidos nucleicos (5,6). Años más tarde, en 1999 se aplicó este principio en el triplete CAG del gen HTT asociado a la enfermedad de Huntington, logrando amplificar por PCR secuencias grandes no metiladas, sin embargo, no se evaluó el efecto de la metilación y su implicancia en el diseño de cebadores, así como los beneficios que ocasionaría la modificación química sobre el microsatélite al facilitar la PCR (7).
El objetivo del estudio fue diseñar una técnica basada en la modificación de las citosinas presentes en el microsatelite CGG del gen FMR1 humano (tripletes CGG) que permita optimizar la reacción de PCR convencional.
MATERIAL Y MÉTODOS
Se seleccionaron aleatoriamente ocho muestras de ADN de voluntarios sanos (cuatro varones y cuatro mujeres) con evaluación clínica normal sin signos de síndrome X frágil realizada por un médico neurólogo, del Instituto Nacional de Ciencias Neurológicas (INCN) obtenidas previamente para estandarización de pruebas de diagnóstico molecular y que se encuentran almacenadas con código numérico arábigo en el Servicio de Neurogenética del instituto. El estudio fue aprobado por el Comité de Ética del INCN.
Preparación de las muestras
Se obtuvieron 200 μL de ADN genσmico por cada muestra, con concentraciones de ADN de entre 1000 a 10000 ng/μL medidas por el sistema de espectrofotometrνa multivolumen-Take Epoch (BioTek, Winooski, VT, USA).
Denaturación del ADN
Según protocolo de Herman y col (8), añadiendo a la denaturación por álcalis original, un paso adicional de denaturación por calor, para acelerar este proceso. A cada muestra de ADN genómico (10000 ng) se le agregó agua bidestilada hasta un volumen final de 18 μL, calentαndola a temperatura de 95°C por 20 minutos en un ciclador de temperatura (GeneAmp® PCR System 9700 Applied. Biosystem, Foster City, CA, USA) y manteniιndola luego a -4°C; posteriormente se agregσ 2 μL de NaOH3M para incubaciσn por 20 minutos a 42°C, obteniιndose un volumen final de 20 μL.
Modificación nucleotídica del ADN
Para cambiar las citosinas por uracilo en las hebras de ADN denaturadas, se utilizó la técnica descrita por Hayatsu y col (9), utilizando mayores concentraciones de iones sulfito para darle celeridad a este proceso. Se agregó 380 μL de una solución 5M de bisulfito de sodio e hidroquinona (pH 5,0). Para evitar la evaporación se agregó seis gotas de aceite mineral, incubándose por 16 h a 50°C. Posteriormente se purificó el ADN modificado utilizando el kit de purificación Zymo Research (10) siguiendo las especificaciones del fabricante; el nivel de purificación se evaluó por electroforesis y espectrofotometría.
Simulación bioinformática en el gen FMR1
Previa caracterización de la estructura de una región de 5180pb del gen FMR1 empleando la base de datos GenBank (11) se realizaron simulaciones de la región promotora y no traducible del gen FMR1 (secuencia de interés de 718pb) con UR de 19, 50, 200 y 2 000 tripletes, así como temperaturas variables que van desde 37°C a 98°C. La simulación de patrones de metilación para la secuencia de interés del gen nativo fue realizada con el programa CpGPlot (12) La simulación de estructuras alternativas que potencialmente adoptaría la secuencia de interés del gen nativo y del gen modificado se realizó utilizando el programa MFOLD (13).
Diseño de cebadores
Se diseñaron cuatro cebadores utilizando el programa MethPrimer (14), dos para que hibriden en la secuencia nucleotídica formada por citosinas y/o uracilos de la región promotora del gen FMR1 (cebadores M) y otros dos que flanqueen el microsatélite que contiene los tripletes CGG de la región no traducible del gen (cebadores T).
Amplificación por PCR
Utilizando PCR convencional y los cebadores diseñados, se amplificó la secuencia de interés del gen FMR1 modificado. La visualización de las amplificaciones se realizó por electroforesis utilizando PAGE al 6% con tinción de nitrato de plata.
RESULTADOS
Se confirmó la modificación nucleotídica de la secuencia de interés mediante espectrofotometría del nucleótido uracilo, y se obtuvo una concentración media de ADN modificado de 185,064 ± 5,108 ng/μL. El análisis de impurezas del ADN modificado, realizado inicialmente por electroforesis, mostró regularidad de las bandas en el gel PAGE luego de la purificación (Figura 1). Este resultado fue corroborado por espectrofotometría que mostró una concentración inicial de sales de 9,625 ± 6,789 ng/μL y una concentración final de 1,408 ± 1,015 ng/μL posterior a la purificación.
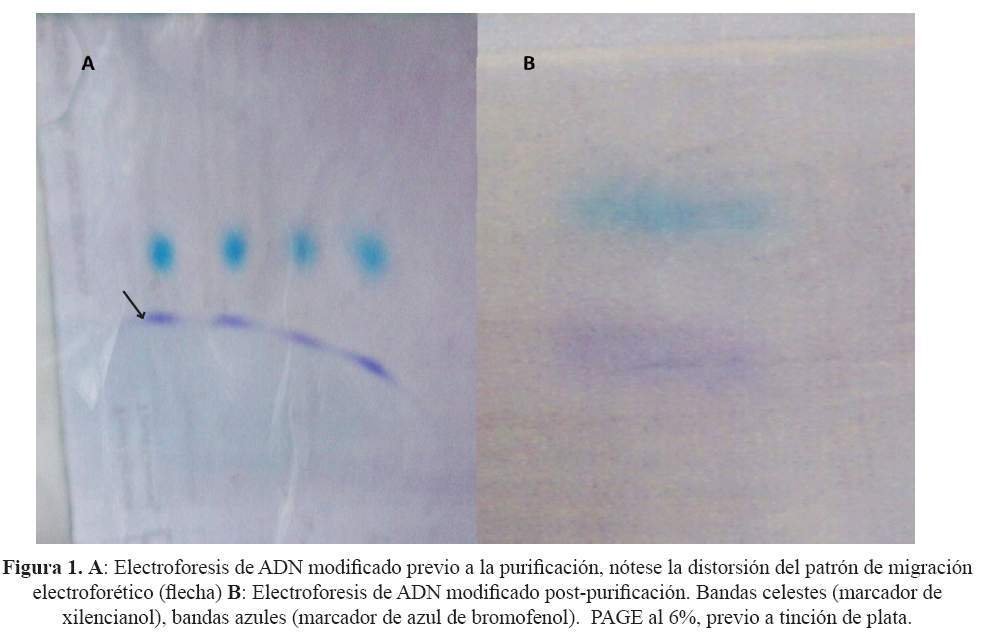
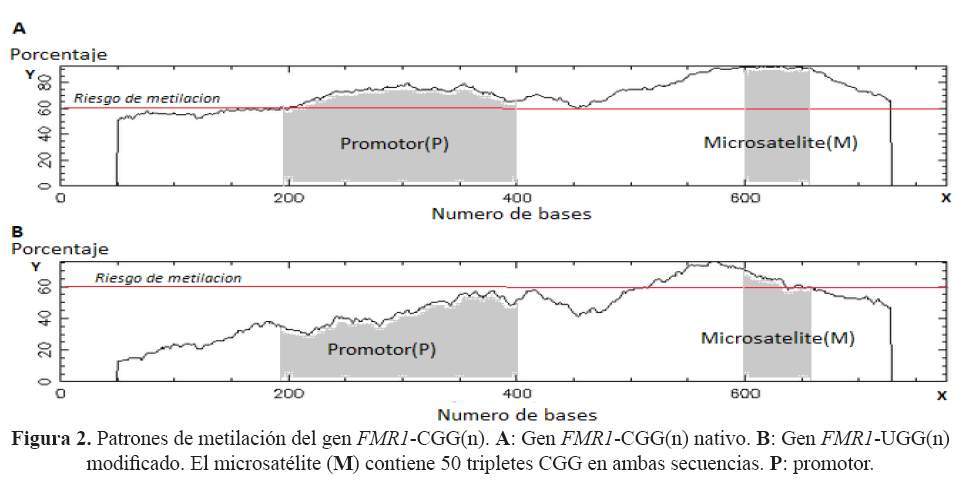
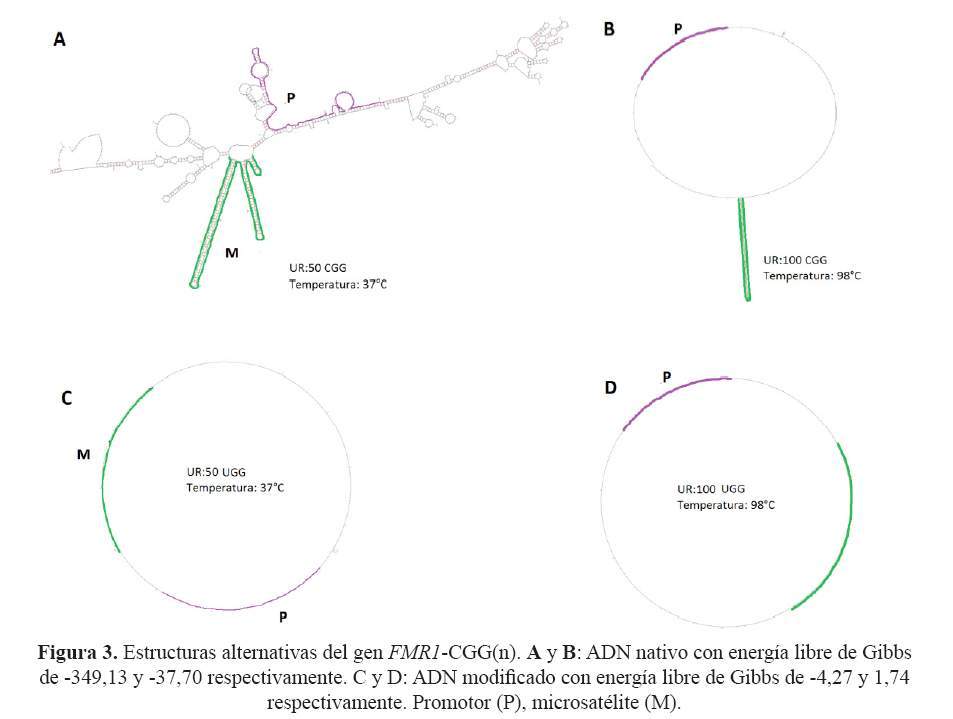
La caracterización bioinformática de la secuencia de interés del gen nativo FMR1 indica que ésta abarca desde el nucleótido -848 hasta el +280 y comprende la potencial isla CpG, el microsatélite y la primera parte de la región traducible. Las simulaciones de metilación y estructura de la secuencia de interés del gen nativo generaron estructuras alternativas y patrones de metilación, mientras que en la condición de ADN modificado generaron ausencia de estos (Figura 2 y figura 3).
Los cebadores T flanquean el microsatélite modificado y toman como molde de amplificación solo una de las cadenas del microsatélite y generan un fragmento de 176pb + (UGG)n. Los cebadores M flanquean la región promotora que contiene citosinas metiladas e hibridan con una secuencia que contiene citosinas y uracilos, generando un amplificado de 143pb. La función de los cebadores M es detectar de manera indirecta la metilación del microsatélite cuando está anormalmente expandido y metilado (Figura 4).
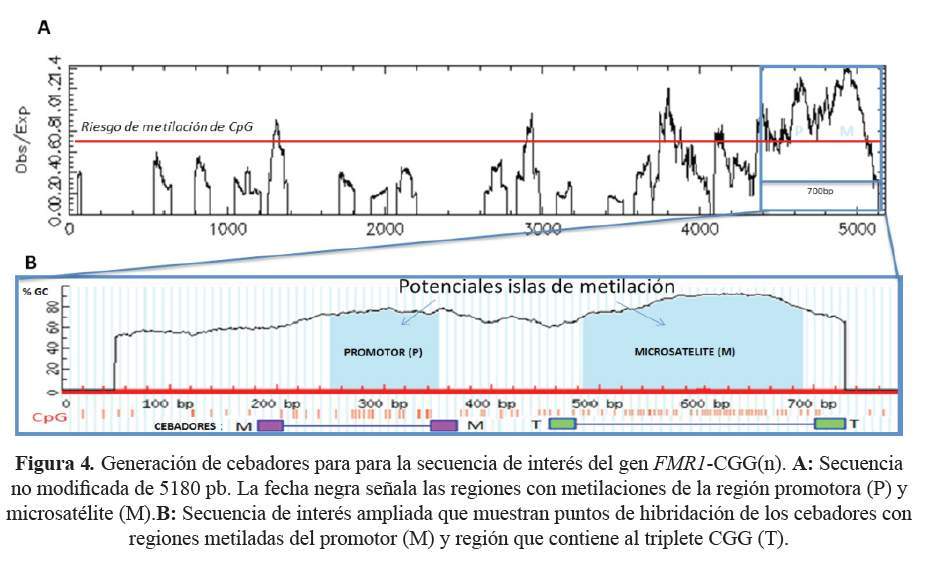
Utilizando los cebadores T, se obtuvieron amplificados de 212pb (12 tripletes UGG) y 221pb (15 tripletes UGG) del ADN, correspondientes a una muestra femenina y masculina respectivamente. Los cebadores M hibridaron en la primera isla CpG metilada de la región promotora generando amplificados de 143 pb para las muestras provenientes de las mujeres participantes del estudio. Por el contrario, en el caso de las muestras de varones normales, no se obtuvo amplificado (Figura 5).
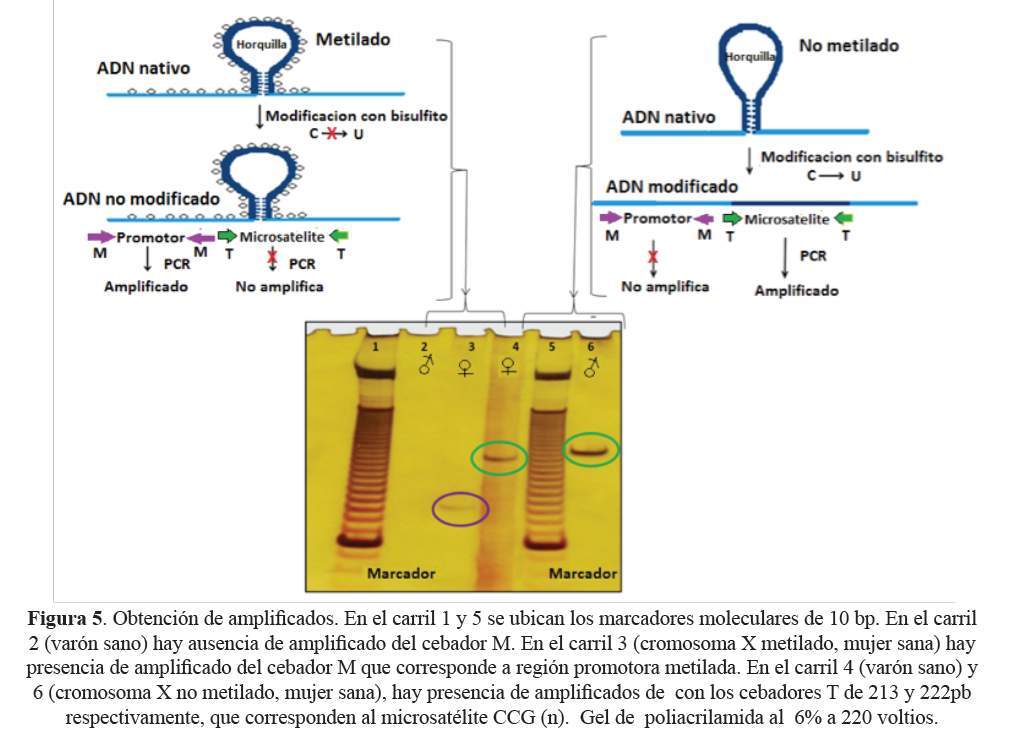
DISCUSIÓN
Este estudio introduce una técnica de modificación química del ADN con bisulfito de sodio y basada en PCR convencional, para genotipar el microsatélite CGG(n) del gen FMR1 relacionado al Síndrome X Frágil.
Luego de la modificación química del ADN genómico se recuperó solo el 10% de la concentración inicial, debido a que el tiempo de exposición al bisulfito necesario para modificación, es prolongado permitiendo la degradación del ADN por reacciones de depurinación, como lo describió Raizis y col (15). La purificación posterior se realizó para eliminar el bisulfito sobrante y el exceso de sales generado por la modificación química y así evitar la inhibición de la reacción de PCR (8).
Se corroboró la posición del microsatelite del gen FMR1 reportada en trabajos previos de Kremer y col(16). La simulación de estructuras mostró que el microsatélite no modificado adopta estructuras en forma de llamadas horquillas que tienden a ser más grandes y complejas a mayor número de UR (Figura 3A). En la simulación a diferentes temperaturas (desde 37° a 98°C) se observó que el MI sigue formando horquillas, esto se debería a la existencia de elementos de simetría y puentes de hidrógeno triples entre las citosinas y guaninas en cada UR generando un proceso de denaturación ineficiente, tal y como los describieron previamente Chong y col (17). Sin embargo, la simulación de la estructura del microsatélite modificado no genera estructuras alternativas, debido a la ausencia de elementos de simetría y a la disminución de puentes de hidrógeno entre ambas cadenas del microsatélite, causada por el reemplazo de citosinas por uracilos (Figura 3 C y D).
Los datos termodinámicos obtenidos de la simulación del triplete muestran la tendencia espontánea (DG<0) a la formación de horquillas del microsatélite no modificado; esto se debería a la gran cantidad de elementos de simetría presentes en el MI, es decir, los tripletes CGG. Ello contrasta con la termodinámica del microsatelite modificado, que mostró una baja tendencia a la formación de horquillas como resultado de la modificación de las citosinas (DG=1.74)(18,19).
En la simulación de la tendencia a metilación en la secuencia no modificada, se encontró dos potenciales islas CpG (Figura 2A). La primera isla CpG está ubicada en el promotor y la segunda en el mismo microsatélite; siendo la segunda isla CpG, un patrón de metilación más notorio a medida que aumentan las UR. Este fenómeno ya ha sido observado in vivo, en el 95% de casos con un silenciamiento del gen FMR1 por metilación (ausencia de la proteína FMRP1), siendo este la base patogénica molecular del Síndrome X Frágil (19,20). La simulación encontró también una tendencia a metilación de microsatélites <200 UR en la región promotora (primera isla) de la secuencia no modificada. In vivo, este fenómeno solo ocurre en menos del 5% de casos de Síndrome X Frágil, esto podría explicarse por errores en el reconocimiento de los puntos CG (invariables en el promotor) por parte de las metilasas y el efecto de las horquillas (21,22). La simulación de la secuencia de interés modificada no evidenció tendencia a metilación de la región promotora ni del microsatelite, esto se explicaría debido la drástica disminución de dinucleótidos CG que impide la formación de islas CpG (Figura 2B).
La obtención final de amplificados con los cebadores T demostró la eficiencia de la modificación del ADN, su purificación, la simulación correcta del microsatélite, así como el diseño adecuado de los cebadores T (Figura 4). Ello también indica que los cambios generados por la modificación química del ADN facilita el proceso de la PCR, ya que el microsatélite modificado está libre de horquillas; otro beneficio de la modificación química es que facilita la denaturación del ADN incrementado su eficiencia con cada ciclo de PCR.
La modificación química del ADN, es decir, la conversión de citosina a uracilo por efecto del bisulfito, no ocurre cuando las citosinas del microsatélite se encuentran metiladas (23). Este fenómeno fue evaluado con los cebadores M, los cuales hibridan en la primera isla CpG metilada de la región promotora. Al obtener amplificados con los cebadores M, en muestras provenientes de las participantes mujeres del estudio, se demostró que la conversión nucleotídica no se dio en el cromosoma X normalmente inactivo por estar metilado (24). La ausencia de amplificado en las muestras proveniente de los participantes varones del estudio, que tienen un cromosoma X activo (desmetilado), evidencia que hubo modificación de ADN por el bisulfito. Ambas observaciones demuestran que los primers M son capaces de discriminar entre regiones metiladas y no metiladas (Figura 4).
La técnica estándar reconocida mundialmente para diagnóstico de Síndrome X frágil es la determinación del número de tripletes CCG por Southern Blot/PCR, esta técnica de hibridización es de muy alto costo, ya que requiere de sondas marcadas con radioactividad o fluorescencia, detectores especiales para la lectura y entrenamiento especializado, además de grandes cantidades de ADN de alta calidad y pureza (26). El diseño propuesto en este estudio requiere menor tiempo para su ejecución, así como menor capacitación del personal y resulta costo-efectivo en comparación a la técnica del Southern Blot/PCR; además, el requerimiento de calidad y cantidad de ADN genómico es de cinco a diez veces menor que la prueba estándar y no requiere uso equipamiento sofisticado haciéndolo de fácil aplicación en nuestro medio (27).
Se han propuesto otros diseños para diagnóstico molecular X frágil como la PCR para alta resolución de metilación "High-resolution methylation PCR" que incluye procesos automatizados que permiten varios análisis en corto tiempo, detecta y cuantifica patrones de metilación y requiere poco ADN de partida; sin embargo requiere sondas marcadas, enzimas de restricción sensibles a metilación y un equipo de electroforesis capilar que elevan sus costos, y son de limitado acceso en nuestro país (28).
Debido a la relación directa entre la metilación de la región promotora y la expansión del microsatétlite, la presencia de amplicones obtenidos por los cebadores M podrían indicar de manera indirecta la presencia de microsatélites metilados (mayores a 200 CGG) (25). La metodología propuesta podría aplicarse para estrategias diagnósticas alternativas del Síndrome X Frágil. Un diagnóstico molecular basado en la modificación química por bisulfito del gen FMR1 permitiría el diagnóstico indirecto y semicuantitativo de personas afectadas por estados de premutación y mutación completa del síndrome X frágil. Para validar este principio, se requiere análisis de muestras de ADN de personas afectadas con el síndrome X Frágil y que hayan sido previamente diagnosticadas por algún procedimiento estándar como Southern blot/PCR.
Aunque los participantes presentaron un examen neurológico normal, no se determinó el número de tripletes CCG del gen FMR1 con la prueba aceptada como estándar a nivel mundial (Southern blot/PCR), pudiendo haberse reclutado participantes, en especial mujeres, con estados premutados del gen FMR1, aunque esta sería una limitación del estudio, no afectaría sus resultados, ya que en los estados de premutación no se encuentra metilación del microsatélite.
En conclusión, la modificación química con bisulfito de sodio del gen FMR1 detecta las secuencias ricas en citosinas metiladas y no metiladas del gen FMR1 en las muestras estudiadas. La evaluación in silico del comportamiento del microsatélite pre y post modificación permitió la evaluación de su estructura primaria y sus tendencia a formar estructuras alternativas, así como el diseño de cebadores específicos para la región promotora (M) y para el microsatélite (T). Este diseño podría ser aplicado, previa validación en pacientes y controles, como alternativa de diagnóstico molecular para el Síndrome X Frágil y otras enfermedades vinculadas a expansión de UR en lugares de difícil aplicación de técnicas costosas y complejas como Southern Blot/PCR.
Agradecimientos
Al PhD Miguel Castro, CEO Bio-Synthesis Inc, USA, por la donación de los cebadores usados en este estudio. Al Dr. João Monteiro de Pina Neto y a la Tec Daniela Dedemo Ribeiro de la Universidad de Sao Paulo, Brasil, por la asesoría temática y entrenamiento molecular en SXF. Este estudio fue apoyado por el NIH Research Training Grant # R25 TW009345 funded the Fogarty International Center, the National Institute of Mental Health, the NIH Office of the Director, Office of Research on Womens Health and the Office of AIDS Research.
Declaración de financiamiento y de conflictos de intereses:
Este estudio recibió financiamiento del Vicerrectorado de la Universidad Nacional Mayor de San Marcos, y del Instituto Nacional de Ciencias Neurológicas. Los autores declaran no tener conflictos de interés.
REFERENCIAS BIBLIOGRÁFICAS
1. Richards RI. Dynamic mutations: a decade of unstable expanded repeats in human genetic disease. Hum Mol Genet. 2001; 10(20):2187. [ Links ]
2. Genç B, Müller-Hartmann H, Zeschnigk M, et al. Methylation mosaicism of 5′-(CGG) n-3′ repeats in fragile X, premutation and normal individuals. Nucleic Acids Res. 2000; 28(10):2141. [ Links ]
3. Filipovic-Sadic S, Sah S, Chen L, et al. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin Chem. 2010; 56(3):399- 408. [ Links ]
4. Liang S, Bass HN, Gao H, Astbury C, Jamehdor MR, Qu Y. A Pseudo-Full Mutation Identified in Fragile X Assay Reveals a Novel Base Change Abolishing an EcoRI Restriction Site. J Mol Diagn. 2008; 10(5):469. [ Links ]
5. Shapiro R, Weisgras JM. Bisulfite-catalyzed transamination of cytosine and cytidine. Biochem Biophys Res Commun. 1970; 40(4):839-43. [ Links ]
6. Hayatsu H, Miura A. The mutagenic action of sodium bisulfite. Biochem Biophys Res Commun. 1970; 39(1):156-60. [ Links ]
7. Panagopoulos I, Lassen C, Kristoffersson U, \AAman P. A novel PCR-based approach for the detection of the Huntington disease associated trinucleotide repeat expansion. Hum Mutat. 1999; 13:232-6. [ Links ]
8. Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci. 1996; 93(18):9821. [ Links ]
9. Hayatsu H, Wataya Y, Kai K, Iida S. Reaction of sodium bisulfite with uracil, cytosine, and their derivatives. Biochemistry (Mosc). 1970; 9(14):2858- 65. [ Links ]
10. Epigenetics, DNA Purification, and RNA Isolation Technologies (Internet). (Citado 4 de julio de 2013). Disponible en: http://www.zymoresearch.com [ Links ] 11. GenBank Home (Internet). (Citado 4 de julio de 2013). URL disponible en:
12. EMBOSS: cpgplot (Internet). (Citado 4 de julio de 2013). URL disponible en: http://emboss.bioinformatics.nl/cgi-bin/emboss/cpgplot [ Links ] 13. DNA folding form | mfold.rit.albany.edu (Internet). (Citado 4 de julio de 2013). URL disponible en: 14. The Li Lab - Tools and Databases. URL disponible en:
15. Raizis AM, Schmitt F, Jost JP. A Bisulfite Method of 5-Methylcytosine Mapping That Minimizes Template Degradation. Anal Biochem. 1995; 226(1):161-6. [ Links ]
16. Kremer E, Pritchard M, Lynch M, Yu S, Holman K, Baker E, et al. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p (CCG) n. Science. 1991; 252(5013):1711. [ Links ]
17. Chong SS, Eichler EE, Nelson DL, Hughes MR. Robust amplification and ethidium-visible detection of the fragile X syndrome CGG repeat using Pfu polymerase. Am J Med Genet. 1994; 51(4):522-6. [ Links ]
18. Sinden RR. Biological implications of the DNA structures associated with disease-causing triplet repeats. Am J Hum Genet. 1999; 64(2):346. [ Links ]
19. Sinden RR, Potaman VN, Oussatcheva EA, Pearson CE, Lyubchenko YL, Shlyakhtenko LS. Triplet repeat DNA structures and human genetic disease: dynamic mutations from dynamic DNA. J Biosci. 2002; 27(1):53-65. [ Links ]
20. Pintado E, Morón F. Metilación y expresión del gen FMR1. Rev Neurol. 2001; 33(1):57-72. [ Links ]
21. Smeets HJM, Smits APT, Verheij CE, Theelen JPG, Willemsen R, Van de Burgt I, et al. Normal phenotype in two brothers with a full FMR1 mutation. Hum Mol Genet. 1995; 4(11):2103-8. [ Links ]
22. Burman RW, Yates PA, Green LD, Jacky PB, Turker MS, Popovich BW. Hypomethylation of an Expanded FMR1 Allele Is Not Associated with a Global DNA Methylation Defect. Am J Hum Genet. 1999; 65(5):1375-86. [ Links ]
23. Hayatsu H. Bisulfite modification of cytosine and 5-methylcytosine as used in epigenetic studies. Genes Environ. 2006; 28(1):1-8. [ Links ]
24. Devys D. Analysts of full fragile X mutations in fetal tissues and monozygotic twins indicate that abnormal methylation and somatic heterogeneity are established early in development. Am J Med Genet. 1992; 43:208 -216. [ Links ]
25. Warren ST. The epigenetics of fragile X syndrome. Cell Stem Cell. 2007; 1(5):488-9. [ Links ]
26. Gold B, Radu D, Balanko A, Chiang CS. Diagnosis of Fragile X syndrome by Southern blot hybridization using a chemiluminescent probe: a laboratory protocol. Mol Diagn. 2000; 5(3):169-78. [ Links ]
27. Nahhas FA, Monroe TJ, Prior TW, et al. Evaluation of the human fragile X mental retardation 1 polymerase chain reaction reagents to amplify the FMR1 gene: testing in a clinical diagnostic laboratory. Genet Test Mol Biomarkers. 2012; 16(3):187-92. [ Links ]
28. Wu D, Zhu Z, Zhao Z, Qu Y, Yang J. Methylation analysis of CpG island DNA of FMR1 gene in the fragile X syndrome. Chin J Med Genet. 2013; 30(1):60-3. [ Links ]
Correspondencia:
Saúl Lindo-Samanamud.
Centro de investigación en Neurogenética, Instituto Nacional de Ciencias Neurológicas. Jirón Ancash 1271 Lima 01, Perú.
Teléfono:(51)1-411-7779
Correo electrónico: saul.lindo.s@incngen.org.pe
Recibido: 14/05/2013
Aceptado: 19/09/2013
