










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Medica Herediana
versión impresa ISSN 1018-130X
Rev Med Hered vol.26 no.2 Lima abr./jun. 2015
Guía para diagnóstico y tratamiento de la Enfermedad de Gaucher
Guidelines for diagnosis and treatment of Gaucher Disease
Maria Colquicocha-Murillo1,2,a;3,b, Janetliz Cucho-Jurado4,a,c, Renee Mercedes Eyzaguirre-Zapata 3,5,6,b;7,d,e, Gioconda Manassero-Morales8,f,g,h, Mariela del Carmen Moreno-Larrea3,9,10,b;11,e,i,j, Katia Liliana Salas-Arbizu12,i, Aimee Margarita Torres-Argandoña9,b;11,a,i, Jesús Olga Vargas-Castro6,b;11,a,i
1 Servicio de Hematología Clínica, Hospital Alberto Sabogal Sologuren. Callao, Perú.
2 Servicio de Hemoterapia y Banco de sangre, Hospital Alberto Sabogal Sologuren. Callao, Perú
3 Facultad de Medicina, Universidad Nacional Mayor de San Marcos. Lima, Perú.
4 Servicio de Gastroenterología, Hepatología y Nutrición Pediátrica, Instituto Nacional de Salud del Niño. Lima, Perú.
5 Facultad de Medicina, Universidad Nacional Federico Villarreal. Lima, Perú.
6 Facultad de Medicina, Universidad Ricardo Palma. Lima, Perú.
7 Servicio de Hematología Clínica, Hospital Guillermo Almenara Irigoyen. Lima, Perú.
8 Servicio de Genética, Instituto Nacional de Salud del Niño-San Borja. Lima, Perú.
9 Facultad de Medicina, Universidad Peruana de Ciencias Aplicadas. Lima, Perú
10 Facultad de Medicina, Universidad San Martin de Porres. Lima, Perú.
11 Servicio de Hematología Clínica, Hospital Nacional Edgardo Rebagliati Martins, EsSALUD. Lima, Perú
12 Servicio de hematología pediátrica, evaluación y tratamiento de pacientes con patología hematológica no Neoplásica, Instituto Nacional de Salud del Niño-Breña. Lima, Perú.
a Médico Asistente; b Docente; c Pediatra Gastroenteróloga; d Médico general; e Jefe; f Pediatra; g Especialista en Endocrinología Pediátrica y Genética; h Responsable del Servicio; i Médico hematólogo; j Especialista en Trasplante de Progenitores Hematopoyéticos.
INTRODUCCIÓN
La enfermedad de Gaucher (EG), debe su nombre por haber sido descrita por Phillipe Gaucher en 1882. Es la enfermedad más frecuente del grupo de las enfermedades de depósito lisosomal comprendidas dentro de los errores innatos del metabolismo (1). La enfermedad de Gaucher se debe a mutaciones en el gen responsable de la síntesis de la enzima lisosomal b-glucocerebrosidasa ácida, también llamada ß-Glucosidasa ácida, (o ß-GA), cuyos locus se ubica en 1q21, es decir en la banda uno de la región 2 del brazo largo del cromosoma 1. El patrón de herencia es autosómico recesivo, es decir que la mutación en éste gen debe darse en estado de homocigocia (2).
La deficiencia enzimática favorece la acumulación de la glucosil-ceramida en los lisosomas de macrófagos (células de Gaucher) y monocitos del sistema monocito macrófago (SMM), llevando a la acumulación de glucocerebrósidos en la médula ósea, el bazo, el hígado, los pulmones, tejido esquelético y en las formas neurológicas en el cerebro, causando daño celular y disfunción orgánica. Como consecuencia, las características de la EG son el desarrollo de síntomas y signos multisistémicos que se establecen de manera crónica y progresiva, tales como: visceromegalias, destrucción ósea y citopenias periféricas (3). El espectro clínico de la enfermedad es altamente heterogéneo, incluyendo adultos asintomáticos en los que se diagnostica la enfermedad de manera incidental, hasta formas muy severas como el hidrops fetalis y formas de severidad variable en la edad adulta, es posible observar esta heterogeneidad incluso en hermanos y también en gemelos (4).
La prevalencia de la EG es de 1/40 000 a 1/60 000 personas, salvo en la población de judíos Ashkenazi, en quienes es cien veces mayor. La frecuencia de portadores (heterocigotos) es de 1/14 en personas con ancestros judíos Ashkenazi y de 1/500 en la población general (5).
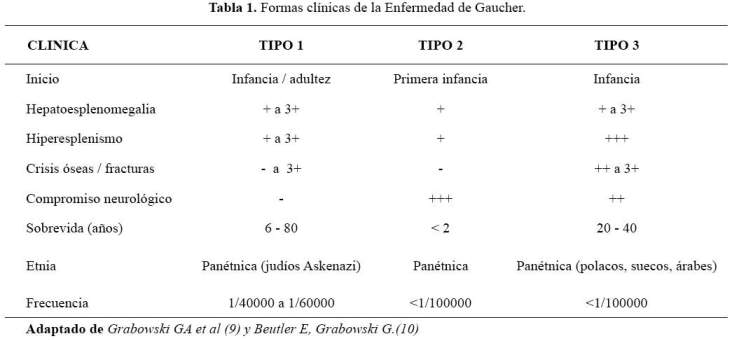
La EG se clasifica en tres formas clínicas ( Tabla 1), en base a la ausencia (tipo 1) o presencia (tipos 2 y 3) de signos neurológicos (1,5). La forma clínica tipo 1 es la más frecuente en todos los grupos poblacionales. Además de las variantes clásicas, se reconocen variantes clínicas infrecuentes como la forma clínica perinatal letal (OMIM # 608013) (6) y la variante cardiovascular (OMIM # 231005) (3,7-9).
El objetivo principal de esta guía es establecer un referente nacional para el diagnóstico, manejo y tratamiento de la Enfermedad de Gaucher, para orientar la toma de decisiones clínicas basadas en recomendaciones sustentadas en la mejor evidencia disponible, ayudando a mejorar el manejo multidisciplinario por parte de los genetistas, hematólogos, internistas, pediatras, y traumatólogos que diagnostican y tratan a pacientes con EG (11).
También son objetivos de esta guía brindar información para estandarizar las acciones a nivel nacional, acerca de:
1. Manifestaciones clínicas que sugieren el diagnóstico de la EG.
2. Establecer los estudios de laboratorio necesarios para corroborar su diagnóstico.
3. Establecer los estudios necesarios para la evaluación inicial de los pacientes con EG tipo 1.
4. Precisar los criterios para iniciar terapia de reemplazo enzimático en los pacientes con EG tipo 1.
5. Especificar la dosis adecuada de la terapia de reemplazo enzimático en los pacientes con EG tipo 1.
6. Establecer los estudios necesarios para el seguimiento de los pacientes con EG tipo 1.
7. Precisar los objetivos terapéuticos que se deben de cumplir con la Terapia de Reemplazo enzimático en los pacientes con EG tipo 1.
8. Especificar los criterios para suspender la Terapia de Reemplazo enzimático en los pacientes con EG tipo 1.
Diagnóstico de la enfermedad de Gaucher
Es importante realizar el diagnóstico precoz de la enfermedad de Gaucher, porque los pacientes se pueden beneficiar de la terapia de reemplazo enzimático (TRE), y prevenir de esta manera el desarrollo de complicaciones irreversibles, como la necrosis avascular articular y el retraso en el crecimiento que tiene implicancias en la calidad de vida (11) ( Tabla 2).

Las manifestaciones clínicas de las esfingolipidosis no se explican únicamente por una deficiencia cuantitativa de enzimas ya que con solo 10-20% de la actividad residual de la enzima es suficiente para llevar una vida normal, por lo que se requiere la interacción de otros mecanismos para que se den estas manifestaciones clínicas (12).
La presentación puede ser en infantes (los de peor pronóstico), jóvenes o adultos; no siempre hay asociación entre la expresión clínica de la enfermedad (fenotipo) y las mutaciones genéticas que se han identificado (7). Entre 10 a 25% de los pacientes son asintomáticos y pueden ser diagnosticados a partir del estudio familiar de un miembro afectado (13,14).
La afectación ósea es la manifestación clínica más frecuente y la que produce mayores secuelas; como parte de la enfermedad ósea, se presenta infiltración de la medula ósea por las células de Gaucher, que pueden estimular la fibrosis, necrosis, infartos, lesiones líticas, osteopenia, deformidades y fracturas patológicas, hechos que se evidencian mediante biopsia de medula ósea y radiológicamente constituyéndose en un método diagnostico (15).
La afectación visceral principalmente del bazo y su crecimiento, correlaciona con la progresión de la enfermedad y sus manifestaciones de citopenia, principalmente trombocitopenia que son documentadas por imágenes y exámenes de sangre (9). La hepatomegalia es menor con excepción de los pacientes esplenectomizados, donde puede haber fibrosis y cirrosis, hasta hematopoyesis extramedular (3).
Los sujetos afectados se distinguen bien de los portadores y los normales por la disminución de la actividad enzimática, constituyendo un método seguro para el diagnóstico de EG (11).
Existen marcadores bioquímicos útiles especialmente para el seguimiento de la actividad de la EG, como la enzima quitotriosidasa que es útil sólo si no existe defecto congénito. Se produce en los macrófagos activados y se incrementa en EG y disminuye cuando hay respuesta al tratamiento con TRE (16).
Otras pruebas útiles son:
− Fosfatasa acida resistente al tartrato, que aumenta en las células de Gaucher.
− Enzima convertidora de angiotensina, los niveles aumentan indicando gravedad de la EG y se usa en déficit de quitotriosidasa.
− Fosfatasa alcalina puede aumentar en pacientes con afectación ósea.
− La resonancia magnética de fémures y columna es útil para la detección de afección esquelética temprana.
El diagnóstico clínico requiere confirmación mediante la detección de ausencia o disminución de la actividad de la enzima B glucocerebrosidasa en plasma, leucocitos, biopsia de tejidos o cultivo de fibroblastos o sangre en papel filtro (4,13,14). En los pacientes con EG, la actividad de la enzima B glucocerebrosidasa es de 0 a 15% de la actividad normal (17).
En portadores heterocigotos puede superponerse la actividad de la enzima, por lo que el método de sangre seca en papel de filtro no es el más apropiado (17).
Manejo y tratamiento de la enfermedad
El pronóstico de la EG cambió radicalmente desde la aparición de la terapia de reemplazo enzimática (TRE) con imiglucerasa. Hasta 1991, la enfermedad era considerada una rareza clínica, cuyo tratamiento era exclusivamente sintomático. En 1974, se demostró que la infusión intravenosa de glucocerebrosidasa placentaria purificada reducía los niveles hepáticos y sanguíneos de glucocerebrósido, pero los resultados no fueron consistentes (18). Posteriormente, y luego de la modificación química de la enzima para dirigirla hacia los macrófagos, se observaron respuestas clínicas objetivas en un estudio piloto realizado por Barton y col (19,20).
Desde su aprobación en 1991 por la FDA, la TRE con imiglucerasa ha modificado el fenotipo de la EG (12). El diagnóstico de la EG en la edad pediátrica, es indicativo de severidad y progresión rápida. La intervención terapéutica temprana con dosis adecuadas de TRE permite, especialmente en niños, evitar la aparición de complicaciones óseas irreversibles (15,19).
Los objetivos más importantes de la TRE se resumen en la tabla 3 (16).

Dosificación de la terapia de reemplazo enzimática
Desde octubre de 2003 luego de una reunión de expertos en Ámsterdam, Holanda, se creó un nuevo y revolucionario concepto en el tratamiento de la enfermedad de Gaucher, las metas terapéuticas. Así, dependiendo del logro o del fracaso de estas metas se establece el concepto de una dosis individual para cada paciente. Si bien el tratamiento con la TRE es de por vida, en la actualidad es posible aumentar o disminuir la TRE de acuerdo a las necesidades individuales de cada paciente (21).
La dosis inicial y la de mantenimiento están en relación con la gravedad del compromiso inicial (riesgo bajo y riesgo alto) y a la respuesta que tiene el paciente a la TRE, es decir, al cumplimiento de las metas terapéuticas (MT) (12).
Al diagnóstico, para decidir la dosis inicial, es indispensable clasificar al paciente por su riesgo. Los niños son considerados siempre de riesgo alto; en cambio, los adultos, que en general tienen una enfermedad menos agresiva, pueden ser de riesgo alto o bajo (21,22).
Por lo tanto el tratamiento de la EG tiene dos etapas:
a) Dosificación inicial: depende del riesgo inicial luego de las evaluaciones al diagnóstico.
b) Dosis posterior (mantenimiento): luego de al menos dos años de terapia inicial a dosis sostenida de acuerdo al riesgo, de acuerdo al logro de las MT se puede disminuir o aumentar la dosis de la TRE
Tipos de tratamiento
1) Terapia de reemplazo enzimático (TRE): Existen tres drogas aprobadas por la FDA: Imiglucerasa, velaglucerasa y taliglucerasa. En nuestro país sólo está disponible comercialmente la imiglucerasa, para su uso en EG.
2) Terapia de reducción del sustrato: Miglustat, eliglustat, fármacos aún no disponibles comercialmente en nuestro país.
El tratamiento de primera línea es la TRE con imiglucerasa.
Criterios de inclusión para el inicio de TRE con imiglucerasa
a) Diagnóstico confirmado de EG tipo 1 mediante la determinación de la actividad de la enzima B-glucocerebrosidasa en plasma, leucocitos, biopsia de tejidos o en cultivos de fibroblastos (21).
b) En pacientes con EG tipo 3, el tratamiento debe ser iniciado precozmente, antes que el cuadro neurológico grave se establezca (17).
c) Una o más de las siguientes manifestaciones:
1. Hemoglobina 2 g/dl por abajo del límite normal para la edad y sexo en al menos dos mediciones realizadas con un mes de diferencia y que se hayan excluido otras causas de anemia.
2. Plaquetas menores de 100 000 mm3 en al menos dos mediciones realizadas con un mes de diferencia.
3. Requerimiento transfusional.
4. Hígado con incremento ≥ 1,25 veces del tamaño normal (por TAC o RMN)
5. Bazo con incremento mayor o igual a 10 veces el tamaño normal (por TAC o RMN)
6. Esplenectomía previa.
7. Enfermedad ósea (Osteopenia, deformidad en matraz de Erlenmeyer, osteoartrosis, osteoesclerosis, osteoporosis, fracturas patológicas, necrosis aséptica, osteolisis, infiltración medular, prótesis articulares, dolor o crisis ósea)
8. Afección pulmonar atribuida a EG (neumopatía intersticial, fibrosis)
9. Niños con retardo en crecimiento pondo estatural (niños con estatura debajo del percentil 3 o disminución en la velocidad del crecimiento.)
10. Síntomas como dolor abdominal, debilidad o caquexia o compromiso de la calidad de vida.
Las recomendaciones para iniciar el tratamiento con TRE y su posterior ajuste de dosis se basan en el seguimiento del paciente. El paciente con EG tipo 1 que en la evaluación inicial no presentó criterios para recibir TRE deberá vigilarse según lo establecido en la tabla 4.
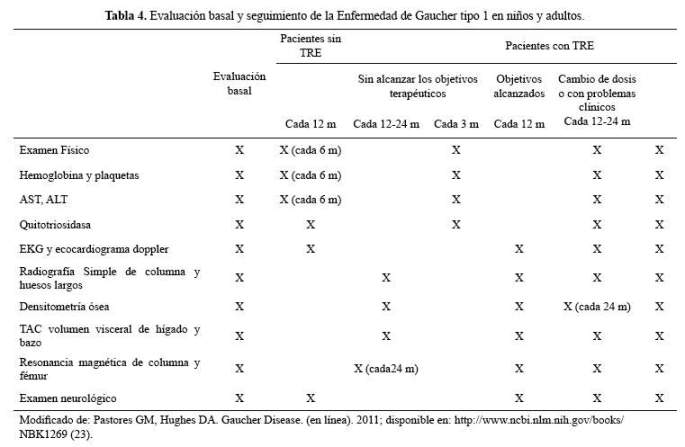
En el caso que el paciente, en algún momento de la evolución cumpla con dichos criterios, se iniciará la TRE (21).
Tratamiento de reemplazo enzimático
Dosis inicial de TRE
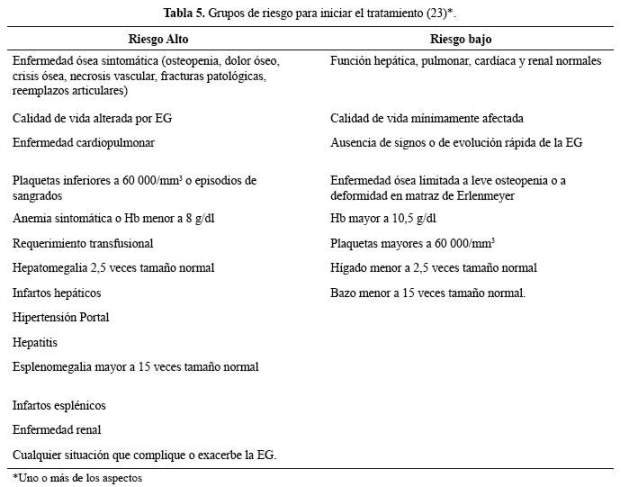
Luego de la evaluación inicial del paciente, se debe diferenciar dos grupos, pacientes de alto y de bajo riesgo ( Tabla 5).
a) Niños y adultos con alto riesgo:
Dosis inicial de 60 U/kg cada 2 semanas, con ajuste posterior (dosis de mantenimiento) de acuerdo con la respuesta clínica y el logro de las metas terapéuticas (MT); los niños son siempre considerados de riesgo alto.
b) Los adultos de riesgo bajo:
Dosis inicial de 30 - 45 U/kg cada 2 semanas, con ajuste posterior (dosis de mantenimiento) de acuerdo con la respuesta clínica y el logro de las MT (24-26).
Dosis de mantenimiento
Podrá aumentar o disminuir, según el logro de las MT de cada paciente (25).
En base a los datos del Registro Internacional de Gaucher se han definido las MT a alcanzar con la TRE. Estas metas se han establecido como guías para los médicos tratantes y deberán ser utilizados anualmente como método para evaluar la respuesta a la TRE y decidir así, cómo continúa la dosificación (25).
La falta de respuesta indica que el paciente necesita una dosis más alta. El objetivo final es determinar y mantener la dosis terapéutica que logre y conserve los beneficios de la terapia en cada paciente.
Los pacientes que requieren TRE deben continuar la terapia por tiempo indefinido (de por vida). La interrupción prolongada de la TRE, especialmente en niños, se asocia con recaída de la enfermedad y mayor compromiso óseo (25). Por lo tanto no son apropiados los períodos prolongados sin terapia (21,27).
Metas terapéuticas
Las MT recomendadas para el paciente con EG tipo I que recibe TRE son (24,26,27):
1. Anemia: Incremento del nivel de hemoglobina en los primeros 12-24 meses y mantener estos valores durante este período: 11g/dl en mujeres y niños, 12 g/dl para hombres mayores de 12 años.
2. Trombocitopenia:
a. Pacientes con esplenectomía: normalización del recuento plaquetario en el primer año de tratamiento.
b. Pacientes con bazo intacto: incremento del recuento plaquetario 1,5 a 2 veces del reporte basal en el primer año y aproximación al nivel normal a partir del segundo año de tratamiento.
3. Hígado: Reducción y mantenimiento del volumen 1,5 veces del valor basal (en función de la superficie corporal del individuo) o reducción en el tamaño en un 20-30% antes de 2 años de tratamiento, entre el 30 y 50% antes de 5 años.
4. Bazo: Reducción y mantenimiento del volumen de 2 a 8 veces el valor basal (en función de la superficie corporal del individuo) o reducción en el tamaño del bazo en un 30-50% antes de un año de tratamiento y entre el 50-60% antes de 5 años.
5. Alteraciones óseas: Disminución o eliminación del dolor óseo en 12 a 24 meses.
a. Mejoría de la mineralización ósea valorada a través de resonancia magnética de columna y fémures después de un año de TRE o mejoría de densidad ósea entre 3 y 5 años.
b. No presentar crisis óseas, osteonecrosis ni aplastamientos subcondrales articulares
6. Biomarcadores: Disminución de la quitotriosidasa del 40 a 70% en los primeros 12 a 24 meses de TRE.
Cambio de dosis en la TRE
Luego de al menos dos años de terapia inicial a dosis sostenida de acuerdo al riesgo, según el logro de las MT se puede disminuir o aumentar la dosis de la TRE (26,27):
− En pacientes adultos de alto riesgo que alcanzaron las MT, se podrá disminuir 15 a 25% de la dosis inicial, durante 3 a 6 meses, con posterior evaluación. La dosis de mantenimiento no debe ser inferior a 30 U/kg cada 2 semanas.
− En pacientes pediátricos se puede disminuir hasta un máximo de 30 U/kg cada 2 semanas
El objetivo final es determinar la dosis adecuada y terapéutica de cada paciente (concepto de dosis individual); si se logran se puede reducir la dosis, si no se logran se debe aumentar la dosis de la TRE.
La TRE es el tratamiento estándar actual que ha mejorado significativamente la calidad de vida de los pacientes con EG Tipo 1(22,26,27).
Los ajustes de la dosis o de la frecuencia de administración sólo deben ser considerados cuando las MT más relevantes han sido alcanzadas, las que deberán mantenerse ante cualquier modificación del régimen terapéutico.
La pérdida de alguna meta alcanzada indica falla en las modificaciones del tratamiento, aunque se debe considerar también la posibilidad de una patología asociada que pudiera confundir el análisis de la evolución de la EG (27).
El tratamiento individualizado de los pacientes con TRE asociada a otros fármacos o medidas terapéuticas, dirigido por un equipo multidisciplinario y especializado en enfermedades metabólicas, es el método más eficiente y de mejor relación costo-beneficio para mejorar la salud de los pacientes con EG Tipo 1 (28).
Para evitar el compromiso óseo se debería iniciar la TRE en forma temprana. La TRE con imiglucerasa ha demostrado mejorar las crisis de dolor y dolor óseo, disminuyendo el riesgo de eventos esqueléticos tales como infarto óseo, lesiones líticas y fracturas, incrementa la DMO de columna lumbar y cuello femoral durante los primeros 4 años de tratamiento (29,30).
Imiglucerasa (26,31)
Modo de administración:
La imiglucerasa es habitualmente administrada por infusión endovenosa periférica. El tratamiento puede ser administrado por vía venosa central en caso de dificultad en acceso venoso periférico (26).
Efectos indeseables:
El tratamiento es generalmente bien tolerado, pero pueden sobrevenir:
Signos generales sin gravedad: diarrea, dolor abdominal, mareos.
Signos de gravedad: manifestaciones cutáneas (urticaria, angioedema, prurito, rash), molestias respiratorias, incluso excepcionalmente shock anafiláctico. Estos signos aparecen esencialmente al inicio del tratamiento, justificando una administración en un centro hospitalario durante los primeros dos años de tratamiento (24).
La duración de la perfusión debe estar adaptada a la posología administrada, inicialmente 2 a 3 horas para 60 U/kg. En ausencia de efectos indeseables, puede realizarse un aumento de la velocidad de la perfusión (una hora y media) (26).
En caso de presentarse algún efecto indeseable menor en el curso de la infusión, se debe reducir la velocidad de infusión, pudiendo llegar a la interrupción temporal de la infusión hasta la desaparición de los síntomas o a la administración de antihistamínicos o de corticoides.
En caso de efecto indeseable semejante a hipersensibilidad, la infusión debe ser interrumpida y un tratamiento apropiado debe ser instaurado. La búsqueda de anticuerpos IgG anti-imiglucerasa (si disponible) debe realizarse en el curso del episodio agudo (25).
En caso de antecedente de reacciones de intolerancia agudas ligeras o moderadas, un pre-tratamiento con antihistamínicos o corticoides, vía oral o vía endovenosa, de 1 a 24 horas antes de la infusión puede evitar reacciones posteriores (31).
Contraindicaciones:
Hipersensibilidad al medicamento o a uno de los excipientes, que ponga en riesgo la vida, es una contraindicación definitiva a la administración de imiglucerasa (31).
Embarazo:
No se cuentan con estudios de la acción de imiglucerasa sobre la reproducción de animales. Se ignora si puede afectar al feto en caso de administración durante embarazo o si puede afectar las funciones de reproducción. No se debería administrar durante la gestación excepto cuando la indicación y necesidad sean claras y el potencial beneficio es juzgado por el médico para sustancialmente justificar el riesgo (32).
Velaglucerasa Alfa (20,30,33-37)
Indicada para la TRE a largo plazo en pacientes con EG tipo 1.
Posología:
La dosis recomendada es 60 U/kg cada 15 días. Pueden hacerse ajustes a la dosis en forma individual, en función del logro y el mantenimiento de los objetivos terapéuticos. En los estudios clínicos se han evaluado dosis de 15 a 60 U/kg cada dos semanas. No se han estudiado dosis superiores a 60 U/kg. A los pacientes actualmente en tratamiento con terapia de sustitución enzimática con imiglucerasa para la enfermedad de Gaucher de tipo 1 puede cambiárseles a VPRIV, utilizando la misma dosis y frecuencia (33).
Método de administración:
Exclusivamente para infusión intravenosa. Deberá administrarse en forma de perfusión intravenosa de 60 minutos de duración. Deberá administrarse a través de un filtro de 0,22 μm.
Contraindicaciones:
Reacción alérgica severa al principio activo o a alguno de los excipientes.
Mujeres con capacidad reproductiva:
Las pacientes con enfermedad de Gaucher que quedan embarazadas pueden experimentar un periodo de exacerbación de la enfermedad durante el embarazo y el puerperio. Es necesaria una evaluación de riesgos y beneficios para cada embarazo. Se requiere una vigilancia estrecha del embarazo y de las manifestaciones clínicas de la enfermedad de Gaucher para la individualización del tratamiento (38).
Embarazo:
No existen datos sobre la utilización de velaglucerasa alfa en mujeres embarazadas. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos sobre el embarazo, el desarrollo embrional/fetal, el parto ni el desarrollo posnatal. Debería prestarse atención en la prescripción a mujeres embarazadas (38).
Lactancia:
No hay datos provenientes de estudios en mujeres en periodo de lactancia. Se desconoce si la velaglucerasa alfa se excreta en la leche materna. Como muchos principios activos se excretan en la leche materna, se debe tener precaución al recetar el medicamento a mujeres en periodo de lactancia.
Fertilidad:
Los estudios en animales no muestran evidencias de deterioro de la fertilidad.
Reacciones adversas:
Reacciones adversas muy frecuentes: cefaleas, mareos, dolor óseo, artralgias, dolor de espalda, reacción relacionada con la perfusión, astenia/fatiga, pirexia/aumento de la temperatura corporal. Reacciones adversas frecuentes: reacción por hipersensibilidad, taquicardia, hipertensión, hipotensión, rubor, dolor abdominal/dolor en la zona superior del abdomen, náuseas, erupción, urticaria (20,33).
Taliglucerasa Alfa (39-44)
Es una enzima lisosomalhidrolítica específica de glucocerebrósido indicada para TRE a largo plazo para adultos con un diagnóstico confirmado de enfermedad de Gaucher tipo 1 (45).
Dosis:
La dosis recomendada es 60 U/kg de peso corporal, administrado una vez cada 2 semanas en infusión intravenosa de 60-120 minutos. Los pacientes actualmente en tratamiento con imiglucerasa para la enfermedad de Gaucher Tipo 1 pueden cambiarse a taliglucerasa. Se recomienda que los pacientes previamente tratados con una dosis estable de imiglucerasa comiencen el tratamiento con taliglucerasa a esa misma dosis. Se pueden hacer ajustes de dosis en función del logro y mantenimiento de los objetivos terapéuticos de cada paciente. Los ensayos clínicos han evaluado rangos de dosis desde 11 a 73 U/kg cada dos semanas.
La velocidad inicial de infusión debe ser 1,3 ml/min. Después de que se haya establecido la tolerancia del paciente a la velocidad de infusión, la misma puede aumentarse a 2,3 ml/min. El volumen total de la solución de infusión debe administrarse durante un período de no menos de una hora (39).
Contraindicaciones:
No se han descrito contraindicaciones hasta el momento.
Uso en poblaciones específicas:
− Embarazo: Categoría B: Se llevaron a cabo estudios de reproducción con taliglucerasa alfa en ratas preñadas a dosis intravenosas de hasta 55 mg/kg/día (alrededor de 5 veces la dosis humana recomendada de 60 U/kg en base a la superficie corporal) y en conejas preñadas a dosis intravenosas de hasta 27,8 mg/kg/día (alrededor de 5 veces la dosis humana recomendada de 60 U/ kg en base a la superficie corporal). Estos estudios no revelaron prueba alguna de deterioro de la fertilidad o daño al feto debido a taliglucerasa alfa. Sin embargo, no existen estudios adecuados y bien controlados en mujeres embarazadas. Debido a que los estudios de reproducción animal no siempre son predictivos de la respuesta humana, taliglucerasa debe utilizarse durante el embarazo sólo si es claramente necesario.
− Lactancia: No hay datos de estudios en mujeres en período de lactancia. No se sabe si este fármaco se excreta en la leche humana. Dado que muchos fármacos se excretan en la leche humana, se debe tener precaución cuando se administra taliglucerasa a una mujer en período de lactancia.
− Uso Pediátrico: La seguridad y eficacia de taliglucerasa en pacientes pediátricos no ha sido establecida. Un paciente pediátrico de 8 años de edad, experimentó una reacción adversa severa (gastroenteritis)(46).
− Uso Geriátrico: Durante los estudios clínicos 8 pacientes de 65 años de edad o mayores fueron tratados con Taliglucerasa. Los estudios clínicos no incluyeron un número suficiente de sujetos de 65 años o más para determinar si responden de manera diferente a personas más jóvenes (45).
Reacciones adversas:
Los síntomas más frecuentemente observados de reacciones a la infusión fueron dolor de cabeza (16%), dolor o molestia en el pecho (6%), astenia (7%), fatiga (5%), urticaria (7%), eritema (5%), presión arterial elevada (5%), dolor de espalda y la artralgia (7%) y
enrojecimiento facial (6%). Otras reacciones alérgicas o a la infusión incluyeron, angioedema, sibilancias, disnea, tos, cianosis e hipotensión. La mayoría de estas reacciones fueron leves y no requirieron tratamiento.
El manejo de las reacciones a la infusión se debe basar en el tipo y severidad de la reacción, por ejemplo, disminuyendo la velocidad de infusión o mediante tratamiento con medicamentos tales como antihistamínicos y antipiréticos (45).
El pre-tratamiento con antihistamínicos o corticosteroides puede prevenir las reacciones posteriores en aquellos casos en que se requiere un tratamiento sintomático. Los pacientes fueron pre-medicados de rutina antes de la infusión de taliglucerasa durante los estudios clínicos (39).
Otras terapias para la enfermedad de Gaucher tipo 1:
Terapia de reducción de sustrato
Mecanismo de acción: inhibición de la Glucocilceramida sintetasa, enzima responsable del primer paso de la síntesis de los glucolípidos disminuyendo así, la producción de glucocerebrósidos (47,48).
Vía de administración: Oral
− Miglustat (49).
− Eliglustat.
a) Miglustat
Reduce la síntesis de glucosilceramida por inhibición reversible de la glucosilceramida sintetasa, enzima responsable del primer paso de la síntesis de los glucolípidos. Fue aprobado por la agencia europea para la evaluación de productos medicinales (EMEA) en 2002 y posteriormente por la USA FDA (2003) (50). Es un derivado sintético de la familia de alcaloides polihidroxilados o imino azúcares extraídos de plantas y microorganismos. El nombre químico de miglustat es 1,5 butilimino-1,5-dideoxi-D-glucitol, tiene un peso molecular de 219 (47).
Indicaciones aprobadas
Pacientes con EG sintomática leve a moderada para quienes la TRE no es apropiada o no constituye una opción terapéutica (FDA). Debe ser indicado y supervisado por médicos con experiencia en EG. Dosis recomendada: 100 mg cada 8 horas vía oral.
No recomendado en menores de 18 años o mayores de 65 años (FDA). Contraindicado en el embarazo, lactancia y en varones y mujeres que desean tener hijos. Tener cautela en pacientes con compromiso de la función renal.
Efectos adversos
Son frecuentes la diarrea y pérdida de peso hasta en un 89% durante el primer año. La diarrea puede deberse a la inhibición de la actividad de la disacaridasa intestinal que resulta en diarrea osmótica (50). Estos síntomas pueden disminuir con una dieta libre de lactosa. La causa de la pérdida de peso se desconoce, su incidencia es mayor en el primer año de tratamiento. Los temblores fueron diagnosticados en 30 % de los pacientes en los primeros meses de terapia. Las parestesias están presentes en el 20% y se resuelven al disminuir a dosis o en forma espontánea (48).
Existen estudios en poblaciones de pacientes adultos comparando la terapia con imiglucerasa y miglustat que reportan mejoría estadísticamente significativa en las MT hematológicas y en las organomegalias con una tasa de respuesta más lenta en comparación de la imiglucerasa. Así mismo reportan mejoría de los síntomas óseos en forma temprana. Sin embargo, Cox (51) en un estudio prospectivo concluyó que el tiempo de seguimiento de los pacientes tratados no predice la estabilidad de la respuesta terapéutica. Otros autores en un estudio con el objetivo de determinar si miglustat lograba mantener las MT alcanzadas por la TRE con imiglucerasa encontraron que el 50% de los pacientes fueron retirados del estudio por efectos adversos y el 75% no logró mantener las MT generales (48).
Son necesarias evidencias a largo plazo en cuanto al tratamiento de las manifestaciones óseas. Las ventajas potenciales deben ser evaluadas cuidadosamente en el contexto de cada paciente.
En conclusión, miglustat es una terapia oral que ha demostrado mejorar algunos aspectos de la EG tipo 1 leve a moderada. Los pacientes adultos con actividad enzimática residual y síntomas leves de la enfermedad pueden ser tratados con miglustat si la TRE no es opción (hipersensibilidad, dificultad con los accesos venosos) (47).
b) Eliglustat
Nueva molécula oral en fase de desarrollo. Su mecanismo de acción es por inhibición parcial de la enzima glucosilceramida sintetasa disminuyendo así, la producción de glucocerebrosidos. La molécula es química y biológicamente diferente del miglustat (51). Fue aprobada por la FDA el 28 de Agosto del 2014.
En un estudio de fase II, después de 4 años de tratamiento con eliglustat (50), 19 pacientes naive con EG tipo I que completaron el tratamiento presentaron un aumento de los niveles de Hb y plaquetas en 2,3 ± 1,5 g/dl y en 68,700 ± 21,200/mm3, respectivamente. Los volúmenes esplénico y hepático disminuyeron en 63% (basal: 17,3 ± 9,5 MN) y en 28% (basal: 1,7 ± 0,4 MN), respectivamente. La quitotriosidasa y el CCL-18 disminuyeron en 82% cada uno; se normalizaron la glucosylceramida y el GM3 (53).
El t-score de la densidad mineral ósea para la espina lumbar aumento en 0,8 (60%). No se reportaron crisis óseas. Los efectos adversos fueron leves y no se relacionaron con el tratamiento. Todos estos resultados muestran seguridad y eficacia de eliglustat que fueron reportados durante 1, 2 y 4 años de tratamiento (51).
Esplenectomía
La esplenectomía es actualmente considerada una práctica de excepción para pacientes con trombocitopenia severa sintomática o con clínica de compresión abdominal (hidronefrosis) aun en pacientes con TRE (53,54).
La esplenectomía, que implica la remoción de un órgano reservorio de macrófagos, genera un fenotipo más severo con compromiso pulmonar, óseo y hepático, y disminuye la expectativa de vida con respecto a los pacientes sin esplenectomía (55,56).
Evitar la esplenectomía aunque pudiera ser necesaria durante hemorragias que comprometan la vida.
Consecuencias de la suspensión de terapia de reemplazo enzimático en la enfermedad de Gaucher tipo 1
La TRE debe ser permanente y de por vida por tratarse de una deficiencia enzimática congénita. Es de gran importancia el tratamiento precoz en el niño pues tendrá efecto terapéutico y preventivo de todas las complicaciones derivadas principalmente del compromiso óseo que podrían ser irreversibles (24,27).
Sin embargo, en el transcurso del tratamiento no es raro que se produzcan interrupciones que son debidas principalmente a la dificultad de obtener el medicamento por diversos factores y a la dificultad en el acceso a las consultas médicas en los servicios de salud, más que a factores relacionados con el medicamento como podrían ser los efectos adversos que son raros, ya que son bien tolerados por la mayoría de los pacientes (26-28).
Actualmente a 20 años de la TRE con imiglucerasa que fue el primer producto obtenido por la tecnología del ADN recombinante para tratamiento de la EG, existen reportes importantes acerca de su seguridad y efectividad; sin embargo, no hay suficientes estudios en relación a los efectos de la suspensión de la TRE por largo plazo por ser pocos casos, además por la dificultad para realizar exámenes de laboratorio y de imágenes para monitorear adecuadamente esta interrupción de tratamiento (24,57).
Otra consecuencia es la disminución de la calidad de vida de los pacientes al reaparecer síntomas que mejoraron con el tratamiento aunque es difícil medir estos, siendo un dato subjetivo (18,25,28).
Como conclusión, el tratamiento de la EG es de por vida. Si bien es cierto que hay pocos estudios en la suspensión TRE, se observa en los pacientes reaparición de los síntomas sobre todo dolores óseos y en el caso de las mujeres sangrado genital anormal (11,59,60). Es de gran importancia la terapia principalmente en niños que están en crecimiento para evitar complicaciones óseas y otras relacionadas a enfermedad progresiva (24,57).
Embarazo y enfermedad de Gaucher
Impacto del embarazo en la EG
El embarazo puede representar una carga fisiológica significativa para las mujeres con EG si no recibe tratamiento (61). El embarazo tiene el potencial de exacerbar manifestaciones de EG, lo que agrava el riesgo de complicaciones durante embarazo, parto y post parto (62). Manifestaciones de condiciones preexistentes (anemia, trombocitopenia), aumento del riesgo de hemorragias o hepatomegalia pueden incrementarse o aparecer nuevos síntomas (61).
El daño óseo puede exacerbarse durante y después del embarazo (63). En los pacientes con antecedente de crisis óseas dolorosas, éstas se exacerban al inicio de gestación (32).
En resumen, la asociación de EG con embarazo lleva a la exacerbación de la enfermedad y con esto a las complicaciones en el embarazo y el puerperio (64).
Impacto de la Enfermedad de Gaucher sobre el embarazo
El embarazo puede complicarse por alteraciones hematológicas y óseas; la anemia, trombocitopenia, hepatoesplenomegalia y el compromiso óseo pueden complicar su evolución (14,32).
El sangrado post parto es la complicación más severa, es debido a alteración de la coagulación y de la función plaquetaria (14). Existe incremento de frecuencia de hemorragia post parto, fiebre puerperal y crisis ósea durante el embarazo (38).
Los problemas óseos de cadera pueden contraindicar el parto vaginal y puede ocurrir más daño durante y después del embarazo (14). La tasa de abortos espontáneos es mayor (25%) en mujeres con EG sin tratamiento.
En gestantes en quienes es necesario realizar parto abdominal (cesárea), el manejo anestésico dependerá del grado de compromiso sistémico. La anestesia regional se ha utilizado en muchas pacientes; pero hay escasa evidencia que permita establecer recomendaciones o protocolos (65). En los casos con indicación de cesárea es necesario después de una evaluación clínica cuidadosa solicitar exámenes de laboratorio para detectar alteraciones bioquímicas, hematológicas, sobre todo anemia, trombocitopenia, trastornos de coagulación y otros con la intención de corregirlos y entrar a un quirófano en adecuadas condiciones (65).
Consejería dirigida a evitar complicaciones durante embarazo y puerperio
Ésta debe ser desde antes de la concepción:
− En una mujer en edad reproductiva con diagnóstico de EG sin tratamiento, es necesario recomendar la postergación del embarazo, mediante medidas de anticoncepción de preferencia con métodos de barrera u hormonales hasta el inicio de la TRE (31,33,49).
− El objetivo de la consejería es optimizar la atención a la paciente durante todo su embarazo.
Tratamiento y manejo de complicaciones durante la gestación
El tratamiento recomendado según diferentes estudios a través del tiempo es la TRE, sin embargo, este tratamiento se encuentra dentro de la categoría C como droga administrada durante embarazo (14).
Los fármacos que han sido estudiados en el embarazo son imiglucerasa, velaglucerasa alfa y taliglucerasa alfa. Imiglucerasa es el producto enzimático de mayor tiempo de uso y el más estudiado, sin haberse demostrado efecto teratogénico; con los otros fármacos tampoco se demostraron efectos adversos, pero tienen poco tiempo de estudios y de uso.
Se ha demostrado disminución del riesgo de abortos espontáneos en mujeres tratadas (1,7%) en comparación con quienes no lo reciben (13,3%); también reducción del riesgo de complicaciones durante post parto (21% en las que no recibieron tratamiento vs 7% en quienes recibieron tratamiento (32,38). No se recomienda la suspensión de la TRE durante la gestación.
La TRE con imiglucerasa antes y durante el embarazo ha demostrado ser beneficiosa reduciendo el riesgo de abortos espontáneos y complicaciones relacionadas a EG, especialmente sangrado durante parto y post parto (31,64).
Actualmente las pautas de la Agencia Europea de medicamentos recomiendan considerar el tratamiento antes de la concepción para un óptimo resultado y para las mujeres que ya lo reciben continuarlo durante la gestación (62).
Lactancia
No se ha demostrado efectos adversos en el producto de la gestación en tratamiento con imiglucerasa. Se han realizado mediciones de concentración de imiglucerasa en leche materna antes de la infusión (0,008 nmol/h/ml) y al final de la infusión (0,119 nmol/h/ml); se encontró pequeñas cantidades de imiglucerasa en la leche materna humana (64).
Enfermedad de Gaucher y Parkinson
Varios reportes han asociado las mutaciones o variantes del gen de la glucocerebrosidasa (GBA) con la Enfermedad de Parkinson (EP)(66,67); otros reportes sugieren que estas mutaciones pueden ser factor de riesgo para el desarrollo del parkinsonismo (68) aún en pacientes heterocigotos (69).
Clínicamente los pacientes con EP heterocigotos con variantes patogénicas de GBA, en promedio son más jóvenes al momento del diagnóstico de EP (67) y se asocian más a cambios cognitivos que los pacientes con parkinsonismo sin variantes GBA (70).
En reportes de parkinsonismo en pacientes con EG tipo 1, muestran que lo característico es un curso clínico severo con aparición temprana de signos neurológicos entre la cuarta a sexta década de la vida, progresión agresiva de los signos y son refractarios a la terapia antiparkinsoniana convencional (71).
Se especula que mutaciones en GBA, aún en heterocigotos, puede ser un factor de riesgo en el desarrollo del parkinsonismo. Esto puede ser por una pérdida de función, donde la actividad enzimática disminuida lleva a un incremento focal de glucosilceramida en regiones cerebrales específicas. El exceso de glucosilceramida en neuronas del hipocampo cultivadas aumenta la liberación de calcio desde los almacenes intracelulares, la densidad del retículo endoplásmico, la sensibilidad a agentes neurotóxicos y la activación del receptor de ryanodina, todo lo cual puede resultar en una disfunción neuronal. No obstante, la amplia mayoría de pacientes con EG o portadores nunca desarrollan signos clínicos de parkinsonismo y aun entre los que si lo hacen, no siempre hay concordancia entre sus hermanos afectados también con EG (68).
Enfermedad de Gaucher y patología oncológica
Como sabemos solamente el crecimiento y desarrollo, tiene una programación molecular en cada una de las células mientras que el proceso de envejecimiento, así como las enfermedades neurodegenerativas, y el desarrollo de cáncer es extremadamente complejo y multifactorial. Probablemente las dos teorías que explican estos fenómenos sean la teoría genética y la teoría de los radicales libres, las cuales igualmente se encuentran estrechamente relacionadas (55,72).
A la luz de los nuevos avances científicos en modelos animales, la EG produce un acumulo intracelular de moléculas que no se pueden metabolizar, y los estudios han demostrado que existe un inusual aumento de neoplasias debido a este fenómeno, sin poder precisarse hasta ahora la causa o mecanismo exacto. Sin embargo se sienta que este acúmulo de moléculas sin degradar dentro de la célula, bajo un mecanismo inflamatorio, contribuiría de manera similar al daño oxidativo producido por los radicales libres al DNA, lípidos y proteínas intracelulares, a un daño estructural del material genético que produciría mutaciones y posterior inicio tanto de problemas degenerativos como de neoplasias (73).
Se reconoce, desde hace algunos años, la posible asociación de algunos tipos de cáncer en pacientes con EG (74-76), siendo el mieloma múltiple (MM) el más común (78,79). Las siguientes neoplasias fueron reportadas asociadas a la EG:
− Neoplasias hematológicas: Mieloma múltiple, Linfomas no Hodgkin, Leucemia linfática crónica, Leucemia aguda, Amiloidosis.
− Neoplasias no hematológicas: Hepatocarcinoma.
Diagnóstico, tratamiento y seguimiento de la enfermedad de Gaucher tipos 2 y 3
La enfermedad de Gaucher tipo 2 y 3 (79) presenta compromiso neurológico, son predominantes en países asiáticos y árabes. Existen 2 únicas asociaciones con las formas neuronopáticas (8,9): tipo IIIB caracterizada por inicio tardío, con enfermedad neurológica altamente variable así como características viscerales variables también (norte de Suecia) y el tipo IIIc (80), una variante cardiaca rara entre árabes palestinos (81). El fenotipo II está asociado con un inicio temprano (3 a 6 meses), severa, enfermedad neurológica rápidamente progresiva y muerte dentro de los primeros 2 años de vida (82).
Diagnóstico de las formas neuronopáticas
Síntomas o signos neurológicos: Apraxia oculomotora (81), afectación bulbar o piramidal, con o sin alteraciones cognitivas; hidrops fetal (46), ictiosis o síndrome regresivo; parálisis de la mirada horizontal supranuclear (patognomónico de compromiso neurológico en Gaucher).
Laboratorio
− Prueba Enzimática: Determinación de la actividad enzimática de la glucocerebrosidasa en leucocitos o fibroblastos con técnicas estandarizadas y acreditadas (Rango normal: 2,1-5,3 umol/l/h)(16). La prueba requiere una muestra de 10cc con EDTA mantenida a temperatura ambiente.
− Diagnóstico molecular: Estudio genético para identificar las mutaciones o alteraciones en la secuencia genética que originan la ausencia de actividad enzimática y que determinan alto riesgo de enfermedad neuronopática (ej: homocigoto para la mutación L444P o D409H) (83).
− Biomarcadores: Actividad de quitotriosidasa (84-89) y la quimoquina CCL-18/PARC (4,90) (en pacientes con deficiencia de la quitotriosidasa) al diagnóstico para evaluar la situación basal del paciente (91-93).
Otras evaluaciones
En sospecha de afectación neurológica aguda: Examen clínico neurológico, movimientos oculares, electroencefalograma, y de requerirse, potenciales evocados auditivos.
En la forma neurológica crónica: además del examen neurológico, examen de movimientos oculares, resonancia magnética nuclear, electroencefalograma, potenciales evocados auditivos y pruebas vestibulares. Es necesario realizar una evaluación psicológica que debiera incluir, coeficiente intelectual, pruebas de atención, memoria, y de apraxia. Además, se recomienda realizar una radiografía de tórax y una tomografía axial computarizada torácica si la radiografía de tórax es anormal (94,95).
Seguimiento
Asegurar el diagnóstico enzimático combinado con el análisis de mutaciones y un historial médico y familiar detallado (16,96).
Consejo genético: Seguimiento de rutina bianual para todos los pacientes tratados niños y adultos no tratados con parámetros inestables; evaluaciones anuales para pacientes adultos con enfermedad estable (7,97).
Inicio de la TRE para pacientes con historia familiar (17,98,99) o genotipo malo con hermanos mayores con manifestaciones severas, se debe considerar la terapia presintomática (100).
La presencia de síntomas en la infancia es sospechosa de enfermedad neuronopática (9,60,101). Todos los niños con síntomas neurológicos secundarios a EG son considerados de alto riesgo para la evolución clínica (102,103).
Tratamiento de la forma crónica neuronopática (tipo III)
Tributarios del tratamiento son:
a) Pacientes con forma crónica neuronopática identificada o tipo III comprobada (13,14,104).
b) Niños con riesgo de desarrollar la forma crónica neuronopática (niños con EG hermanos de niños con tipo III comprobada)
c) Niños con genotipos de riesgo y;
d) Niños con inicio de la enfermedad antes de los 2 años de edad y síntomas clínicos graves (21).
La TRE es el único tratamiento farmacológico autorizado, con resultados poco efectivos en la mayoría de los casos, pero en pacientes con esta forma clínica deberán beneficiarse de un intento terapéutico (105).
Puede considerarse el trasplante de médula ósea (106) o de células de cordón procedentes de donante no emparentado cuando no se obtenga buena evolución con la TRE en estos pacientes.
Dosis de imiglucerasa
Forma crónica neuronopática identificada: Iniciar 120U/kg/15 días; si la patología neurológica progresa, pasar a 240 U/kg/15 días durante 6 meses como máximo. Si no hay mejoría, disminuir la dosis a un nivel que permita controlar los síntomas viscerales (26).
Niños en riesgo para desarrollar la forma crónica neuronopática: iniciar con 60 U/kg/15 días y vigilar la evolución por si aparecen síntomas de alteración neurológica orgánica o funcional. Vigilar especialmente la normalidad de los movimientos sacádicos (101).
Tratamiento de la forma aguda neuronopática (tipo II)
No está indicada la TRE (13,14). Puede ensayarse el tratamiento específico con el objetivo de mejorar su calidad de vida. A efectos prácticos, es conveniente diferenciar entre los que tienen afectación piramidal y los que no la tienen (82).
− Sin afectación piramidal y predominio de patología bulbar (estridor, dificultad para la deglución); probar con 120 U/kg cada 2 semanas; revisar dosis y la continuidad del tratamiento a los 6 meses de su inicio.
− Pacientes con afectación piramidal (opistótonos, espasticidad, trismo) y afectación cognitiva importante: ensayar una dosis de 15 U/kg cada 2 semanas solamente para mejorar la visceromegalia.
En niños con enfermedad neuropática, especialmente en formas agudas, no debe insistirse de forma indefinida en el tratamiento, decidiendo conjuntamente con la familia la retirada del mismo después de un tiempo prudencial (6 a 12 meses) sin resultados terapéuticos satisfactorios (101).
Seguimiento:
El seguimiento de los niños con esta patología debe ser individualizado, pero el uso de un protocolo con parámetros mínimos, facilita el control adecuado de los pacientes a largo plazo (14,60,101).
En los niños con enfermedad neurológica debe practicarse un seguimiento específico de esta patología, con independencia de los controles que puedan ser comunes con los otros pacientes.
El monitoreo de pacientes con enfermedad neuronopática crónica debe comprender un examen neurológico exhaustivo por lo menos cada 6 meses.
REFERENCIAS BIBLIOGRÁFICAS
1. Bohra V, Nair V. Gauchers disease. Indian J Endocrinol Metab. 2011; 15(3):182-6. [ Links ]
2. Yildiz Y, Hoffmann P, VomDahl S, et al. Functional and genetic characterization of the non-lysosomal glucosylceramidase 2 as a modifier for Gaucher disease. Orphanet J Rare Dis. 2013; 8:151. [ Links ]
3. Jmoudiak M, Futerman AH. Gaucher disease: pathological mechanisms and modern management. Br J Haematol. 2005; 129(2):178-88. [ Links ]
4. Van Breemen MJ, Bleijlevens B, de Koster CG, Aerts JMFG. Limitations in quantitation of the biomarker CCL18 in Gaucher disease blood samples by surface-enhanced laser desorption/ionization time-of- flight mass spectrometry. Biochim Biophys Acta. 2006; 1764(10):1626-32. [ Links ]
5. Valle D, Beaudet A, Vogelstein B, et al. (Editores). The online metabolic and molecular bases of inherited disease. New York: The McGraw-Hill Companies; 2001 (Citado 6 de octubre de 2014). Disponible en: http://ommbid.mhmedical.com/book.aspx?bookID=474
6. Eblan MJ, Goker-Alpan O, Sidransky E. Perinatal lethal Gaucher disease: a distinct phenotype along the neuronopathic continuum. Fetal Pediatr Pathol. 2005; 24(4-5):205-22. [ Links ]
7. Germain DP. Gauchers disease: a paradigm for interventional genetics. Clin Genet. 2004; 65(2):77-86. [ Links ]
8. Goker-Alpan O, Schiffmann R, Park JK, Stubblefield BK, Tayebi N, Sidransky E. Phenotypic continuum in neuronopathic Gaucher disease: an intermediate phenotype between type 2 and type 3. J Pediatr. 2003; 143(2):273-6. [ Links ]
9. Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis and assessment, consensus statements. Eur J Pediatr. 2004; 163(2):58-66. [ Links ]
10. Beutler E, Grabowski G. Gaucher disease. In: Scriver CR, Baudet AL, Sly WS, Valle D (Eds). The Metabolic & Molecular bases of inherited disease. 8th. New York: Ed. McGraw-Hill; 2001. p. 3635. [ Links ]
11. Zimran A. How I treat Gaucher disease. Blood. 2011; 118(6):1463-71. [ Links ]
12. Giraldo P. Guía de actuación en pacientes con enfermedad de Gaucher tipo 1. Med Clínica. 2011; 137:55-60. [ Links ]
13. Franco-Ornelas S; Grupo de expertos de enfermedad de Gauche. Consenso Mexicano de Enfermedad de Gaucher. Rev Med Inst Mex Seguro Soc. 2010; 48(2):167- 186. [ Links ]
14. Drelichman G, Basack N, Fernandez N, et al. Consenso para la Enfermedad de Gaucher: Grupo Argentino de diagnóstico y tratamiento de la Enfermedad de Gaucher. Hematol. 2013; 17:25-60. [ Links ]
15. Bembi B, Ciana G, Mengel E, Terk MR, Martini C, Wenstrup RJ. Bone complications in children with Gaucher disease. Br J Radiol. 2002; 75(S1): A37-44. [ Links ]
16. Barranger JA, Rice E, Sakallah SA, Sansieri C, Mifflin TE, Cooper DL. Enzymatic and molecular diagnosis of Gaucher disease. Clin Lab Med. 1995; 15(4):899- 913. [ Links ]
17. Sibille A, Eng CM, Kim SJ, Pastores G, Grabowski GA. Phenotype-genotype correlations in Gaucher disease type I: clinical and therapeutic implications. Am J Hum Genet. 1993; 52(6):1094-101. [ Links ]
18. Brady RO, Pentchev PG, Gal AE, Hibbert SR, Dekaban AS. Replacement therapy for inherited enzyme deficiency. Use of purified glucocerebrosidase in Gauchers disease. N Engl J Med. 1974; 291(19):989-93. [ Links ]
19. Barton NW, Furbish FS, Murray GJ, Garfield M, Brady RO. Therapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher disease. Proc Natl Acad Sci. 1990; 87(5):1913-6. [ Links ]
20. Gonzalez DE, Turkia HB, Lukina EA, et al. Enzyme replacement therapy with velaglucerase alfa in Gaucher disease: Results from a randomized, double-blind, multinational, phase 3 study. Am J Hematol. 2013; 88(3):166-71. [ Links ]
21. Martins AM, Valadares ER, Porta G, et al. Recommendations on diagnosis, treatment, and monitoring for Gaucher disease. J Pediatr. 2009; 155(S4):S10-8. [ Links ]
22. Weinreb NJ, Aggio MC, Andersson HC, et al. Gaucher disease type 1: revised recommendations on evaluations and monitoring for adult patients. Semin Hematol. 2004; 41(S5):15-22. [ Links ]
23. Pastores GM, Hughes DA. Gaucher disease. Seattle, WA: University of Washington; 2011. (Citado 26 de noviembre del 2014) Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK1269
24. Charrow J, Andersson HC, Kaplan P, et al. Enzyme replacement therapy and monitoring for children with type 1 Gaucher disease: consensus recommendations. J Pediatr. 2004; 144(1):112-20. [ Links ]
25. Andersson HC, Charrow J, Kaplan P, et al. Individualization of long-term enzyme replacement therapy for Gaucher disease. Genet Med Off J Am Coll Med Genet. 2005; 7(2):105-10. [ Links ]
26. Grabowski GA, Kacena K, Cole JA, et al. Dose-response relationships for enzyme replacement therapy with imiglucerase/alglucerase in patients with Gaucher disease type 1. Genet Med Off J Am Coll Med Genet. 2009; 11(2):92-100. [ Links ]
27. Pastores GM, Weinreb NJ, Aerts H, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol. 2004; 41(S5):4-14. [ Links ]
28. Connock M, Burls A, Frew E, et al. The clinical effectiveness and cost-effectiveness of enzyme replacement therapy for Gauchers disease: a systematic review. Health Technol Assess Winch Engl. 2006; 10(24): 136. [ Links ]
29. Maas M, van Kuijk C, Stoker J, et al. Quantification of bone involvement in Gaucher disease: MR imaging bone marrow burden score as an alternative to Dixon quantitative chemical shift MR imaging-- initial experience. Radiology. 2003; 229(2):554-61. [ Links ]
30. Elstein D, Foldes AJ, Zahrieh D, et al. Significant and continuous improvement in bone mineral density among type 1 Gaucher disease patients treated with velaglucerase alfa: 69-month experience, including dose reduction. Blood Cells Mol Dis. 2011; 47(1):56- 61. [ Links ]
31. Weinreb N, Barranger J, Packman S, et al. Imiglucerase (Cerezyme) improves quality of life in patients with skeletal manifestations of Gaucher disease. Clin Genet. 2007; 71(6):576-88. [ Links ]
32. Zimran A, Morris E, Mengel E, et al. The female Gaucher patient: the impact of enzyme replacement therapy around key reproductive events (menstruation, pregnancy and menopause). Blood Cells Mol Dis. 2009; 43(3):264-88. [ Links ]
33. Morris JL. Velaglucerase alfa for the management of type 1 Gaucher disease. Clin Ther. 2012; 34(2):259- 71. [ Links ]
34. Aerts JMFG, Yasothan U, Kirkpatrick P. Velaglucerase alfa. Nat Rev Drug Discov. 2010; 9(11):837-8. [ Links ]
35. Zimran A, Altarescu G, Philips M, et al. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood. 2010; 115(23):4651-6. [ Links ]
36. Zimran A. Velaglucerase alfa: a new option for Gaucher disease treatment. Drugs Today Barc Spain 2011; 47(7):515-29. [ Links ]
37. van Dussen L, Cox T, Hendriks E, et al. Effects of switching from a reduced dose imiglucerase to velaglucerase in type 1 Gaucher disease: clinical and biochemical outcomes. Haematol. 2012; 97(12): 1850-1854. [ Links ]
38. Elstein D, Hughes D, Goker-Alpan O, et al. Outcome of pregnancies in women receiving velaglucerase alfa for Gaucher disease. J Obstet Gynaecol Res. 2014; 40(4):968-75. [ Links ]
39. Zimran A, Brill-Almon E, Chertkoff R, et al. Pivotal trial with plant cell-expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood. 2011; 118(22):5767-73. [ Links ]
40. Grabowski GA, Golembo M, Shaaltiel Y. Taliglucerase alfa: an enzyme replacement therapy using plant cell expression technology. Mol Genet Metab. 2014; 112(1):1-8. [ Links ]
41. Haddley K. Taliglucerase alfa for the treatment of Gauchers disease. Drugs Today Barc Spain 1998. 2012; 48(8):525-32. [ Links ]
42. Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid. 2012; 7:15-20. [ Links ]
43. Pastores GM, Petakov M, Giraldo P, et al. A Phase 3, multicenter, open-label, switchover trial to assess the safety and efficacy of taliglucerase alfa, a plant cell-expressed recombinant human glucocerebrosidase, in adult and pediatric patients with Gaucher disease previously treated with imiglucerase. Blood Cells Mol Dis. 2014; 53(4):253-60. [ Links ]
44. Van Dussen L, Zimran A, Akkerman EM, et al. Taliglucerase alfa leads to favorable bone marrow responses in patients with type I Gaucher disease. Blood Cells Mol Dis. 2013; 50(3):206-11. [ Links ]
45. Giraldo P, Latre P. Current treatment for Gauchers disease and new prospects. Med Clínica. 2011; 137 (S1):50-4. [ Links ]
46. Stone DL, Sidransky E. Hydrops fetalis: lysosomal storage disorders in extremis. Adv Pediatr. 1999; 46:409-40. [ Links ]
47. Hollak CEM, Hughes D, van Schaik IN, Schwierin B, Bembi B. Miglustat (Zavesca) in type 1 Gaucher disease: 5-year results of a post-authorisation safety surveillance programme. Pharmacoepidemiol Drug Saf. 2009; 18(9):770-7. [ Links ]
48. Pastores GM, Giraldo P, Chérin P, Mehta A. Goal- oriented therapy with miglustat in Gaucher disease. Curr Med Res Opin. 2009; 25(1):23-37. [ Links ]
49. Ficicioglu C. Review of miglustat for clinical management in Gaucher disease type 1. Ther Clin Risk Manag. 2008; 4(2):425-31. [ Links ]
50. Amiri M, Naim HY. Long term differential consequences of miglustat therapy on intestinal disaccharidases. J Inherit Metab Dis. 2014; 37(6): 929-37
51. Cox TM. Eliglustat tartrate, an orally active glucocerebroside synthase inhibitor for the potential treatment of Gaucher disease and other lysosomal storage diseases. Curr Opin Investig Drugs Lond Engl. 2010; 11(10):1169-81. [ Links ]
52. Lukina E, Watman N, Dragosky M, et al. Eliglustat, an investigational oral therapy for Gaucher disease type 1: Phase 2 trial results after 4years of treatment. Blood Cells Mol Dis. 2014; 53(4):274-6. [ Links ]
53. Adas M, Adas G, Karatepe O, Altiok M, Ozcan D. Gauchers disease diagnosed by splenectomy. North Am J Med Sci. 2009; 1(3):134-6. [ Links ]
54. Addison OL, Pritchard H. Specimen of the Spleen from a Case of Gaucher Splenomegaly. Proc R Soc Med. 1924; 17:9-21. [ Links ]
55. Grabowski GA, Saal HM, Wenstrup RJ, Barton NW. Gaucher disease: a prototype for molecular medicine. Crit Rev Oncol Hematol. 1996; 23(1):25-55. [ Links ]
56. Thomas AS, Mehta A, Hughes DA. Gaucher disease: haematological presentations and complications. Br J Haematol. 2014; 165(4):427-40. [ Links ]
57. Drelichman G, Ponce E, Basack N, et al. Clinical consequences of interrupting enzyme replacement therapy in children with type 1 Gaucher disease. J Pediatr. 2007; 151(2):197-201. [ Links ]
58. Cabrera-Salazar MA, ORourke E, Henderson N, Wessel H, Barranger JA. Correlation of surrogate markers of Gaucher disease. Implications for long- term follow up of enzyme replacement therapy. Clin Chim Acta Int J Clin Chem. 2004; 344(1-2):101-7. [ Links ]
59. Martins AM, Lobo CL, Sobreira EAP, et al. Gaucher disease treatment: a Brazilian consensus. Rev Bras Hematol E Hemoter. 2003; 25(2):89-95. [ Links ]
60. Dalmau J, Baldellou A. Guía clínica para el diagnóstico, tratamiento y seguimiento de la enfermedad de Gaucher en la infancia. Acta Pediatr Esp. 2006; 64:175-80. [ Links ]
61. Malinová V, Poupetová H, Dvoráková L, Zeman J. Enzyme replacement therapy for Gaucher disease in twin pregnancy. Int J Gynaecol Obstet Off Organ Int Fed Gynaecol Obstet. 2009; 106(1):64-6. [ Links ]
62. Granovsky-Grisaru S, Belmatoug N, vom Dahl S, Mengel E, Morris E, Zimran A. The management of pregnancy in Gaucher disease. Eur J Obstet Gynecol Reprod Biol. 2011; 156(1):3-8. [ Links ]
63. Fasouliotis SJ, Ezra Y, Schenker JG. Gauchers disease and pregnancy. Am J Perinatol. 1998; 15(5):311-8. [ Links ]
64. Sekijima Y, Ohashi T, Ohira S, Kosho T, Fukushima Y. Successful pregnancy and lactation outcome in a patient with Gaucher disease receiving enzyme replacement therapy, and the subsequent distribution and excretion of imiglucerase in human breast milk. Clin Ther. 2010; 32(12):2048-52. [ Links ]
65. Pinto J, Segura E, Ribeiro S, Amaral R. Manejo anestésico en embarazada con enfermedad de Gaucher tipo I. Rev Esp Anestesiol Reanim. 2010; 57(4):243- 4. [ Links ]
66. Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinsons disease. N Engl J Med. 2009; 361(17):1651-61. [ Links ]
67. Mitsui J, Mizuta I, Toyoda A, et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol. 2009; 66(5):571-6. [ Links ]
68. Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab. 2004; 81(1):70-3. [ Links ]
69. Goker-Alpan O, Schiffmann R, LaMarca M, Nussbaum R, McInerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet. 2004; 41(12):937-40. [ Links ]
70. Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012; 11(11):986-98. [ Links ]
71. Neudorfer O, Giladi N, Elstein D, et al. Occurrence of Parkinsons syndrome in type I Gaucher disease. QJM Mon J Assoc Physicians. 1996; 89(9):691-4. [ Links ]
72. Cleeter MWJ, Chau KY, Gluck C, et al. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem Int. 2013; 62(1):1-7. [ Links ]
73. Pavlova EV, Wang SZ, Archer J, et al. B cell lymphoma and myeloma in murine Gauchers disease. J Pathol. 2013; 231(1):88-97. [ Links ]
74. Shiran A, Brenner B, Laor A, Tatarsky I. Increased risk of cancer in patients with gaucher disease. Cancer. 1993; 72(1):219-24. [ Links ]
75. Zimran A, Liphshitz I, Barchana M, Abrahamov A, Elstein D. Incidence of malignancies among patients with type I Gaucher disease from a single referral clinic. Blood Cells Mol Dis. 2005; 34(3):197-200. [ Links ]
76. Rosenbloom BE, Weinreb NJ, Zimran A, Kacena KA, Charrow J, Ward E. Gaucher disease and cancer incidence: a study from the Gaucher Registry. Blood. 2005; 105(12):4569-72. [ Links ]
77. Mistry PK, Weinreb NJ, Brady RO, Grabowski GA. Gaucher disease: resetting the clinical and scientific agenda. Am J Hematol. 2009; 84(4):205-7. [ Links ]
78. De Fost M, Vom Dahl S, Weverling GJ, et al. Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cells Mol Dis. 2006; 36(1):53-8. [ Links ]
79. Sidransky E, Fartasch M, Lee RE, et al. Epidermal abnormalities may distinguish type 2 from type 1 and type 3 of Gaucher disease. Pediatr Res. 1996; 39(1):134-41. [ Links ]
80. Shah S, Misri A, Bhat M, Maheshwari S. Gauchers disease type III C: Unusual cause of intracardiac calcification. Ann Pediatr Cardiol. 2008; 1(2):144-6. [ Links ]
81. Bohlega S, Kambouris M, Shahid M, Al Homsi M, Al Sous W. Gaucher disease with oculomotor apraxia and cardiovascular calcification (Gaucher type IIIC). Neurology. 2000; 54(1):261-3. [ Links ]
82. Stone D, Carey W, Christodoulou J, et al. Type 2 Gaucher disease: the collodion baby phenotype revisited. Arch Dis Child Fetal Neonatal Ed. 2000; 82(2):163-6. [ Links ]
83. Beutler E, Gelbart T, Scott CR. Hematologically important mutations: Gaucher disease. Blood Cells Mol Dis. 2005; 35(3):355-64. [ Links ]
84. Barone R, Sotgiu S, Musumeci S. Plasma chitotriosidase in health and pathology. Clin Lab. 2007; 53(5-6):321-33. [ Links ]
85. Czartoryska B, Tylki-Szymanska A, Gorska D. Serum chitotriosidase activity in Gaucher patients on enzyme replacement therapy (ERT). Clin Biochem. 1998; 31(5):417-20. [ Links ]
86. Grace ME, Balwani M, Nazarenko I, Prakash-Cheng A, Desnick RJ. Type 1 Gaucher disease: null and hypomorphic novel chitotriosidase mutations-implications for diagnosis and therapeutic monitoring. Hum Mutat. 2007; 28(9):866-73. [ Links ]
87. Guo Y, He W, Boer AM, et al. Elevated plasma chitotriosidase activity in various lysosomal storage disorders. J Inherit Metab Dis. 1995; 18(6):717-22. [ Links ]
88. Hollak CE, van Weely S, van Oers MH, Aerts JM. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J Clin Invest. 1994;93(3):1288-92. [ Links ]
89. Korolenko TA, Zhanaeva SY, Falameeva OV, et al. Chitotriosidase as a marker of macrophage stimulation. Bull Exp Biol Med. 2000; 130(10):948-50. [ Links ]
90. Boot RG, Verhoek M, de Fost M, et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: a novel surrogate marker for assessing therapeutic intervention. Blood. 2004; 103(1):33-9. [ Links ]
91. Mayeux R. Biomarkers: Potential uses and limitations. NeuroRx. 2004; 1(2):182-8. [ Links ]
92. Cox TM. Biomarkers in lysosomal storage diseases: a review. Acta Paediatr Oslo Nor. 1992; 94(S447):39-42. [ Links ]
93. Rolfs A, Giese AK, Grittner U, et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-jewish, caucasian cohort of gaucher disease patients. PLoS ONE. 2013; 8(11):e79732. [ Links ]
94. Kerem E, Elstein D, Abrahamov A, et al. Pulmonary function abnormalities in type I Gaucher disease. Eur Respir J. 1996; 9(2):340-5. [ Links ]
95. Mistry PK, Sirrs S, Chan A, et al. Pulmonary hypertension in type 1 Gauchers disease: genetic and epigenetic determinants of phenotype and response to therapy. Mol Genet Metab. 2002; 77(1-2):91-8. [ Links ]
96. Chamoles NA, Blanco M, Gaggioli D, Casentini C. Gaucher and Niemann-Pick diseases-enzymatic diagnosis in dried blood spots on filter paper: retrospective diagnoses in newborn-screening cards. Clin Chim Acta Int J Clin Chem. 2002; 317(1):191- 7. [ Links ]
97. Giraldo P, Pocoví M, Pérez-Calvo J, Rubio- Félix D, Giralt M. Report of the Spanish Gauchers disease registry: clinical and genetic characteristics. Haematologica. 2000; 85(8):792-9. [ Links ]
98. Whitfield PD, Nelson P, Sharp PC, et al. Correlation among genotype, phenotype, and biochemical markers in Gaucher disease: implications for the prediction of disease severity. Mol Genet Metab. 2002; 75(1):46-55. [ Links ]
99. Orvisky E, Park JK, LaMarca ME, et al. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: correlation with phenotype and genotype. Mol Genet Metab. 2002; 76(4):262-70. [ Links ]
100. Cormand B, Harboe TL, Gort L, et al. Mutation analysis of Gaucher disease patients from Argentina: high prevalence of the RecNciI mutation. Am J Med Genet. 1998; 80(4):343-51. [ Links ]
101. Vellodi A, Bembi B, de Villemeur TB, et al. Management of neuronopathic Gaucher disease: a European consensus. J Inherit Metab Dis. 2001; 24(3):319-27. [ Links ]
102. Shehi B, Bocari G, Vyshka G, Xhepa R, Alushani D. Gauchers Disease in Albanian Children: Casuistics and Treatment. Iran J Pediatr. 2011; 21(1):1-7. [ Links ]
103. Sidransky E, Sherer DM, Ginns EI. Gaucher disease in the neonate: a distinct Gaucher phenotype is analogous to a mouse model created by targeted disruption of the glucocerebrosidase gene. Pediatr Res. 1992; 32(4):494-8. [ Links ]
104. Botross NP, Riad AA, Viswanathan S, Nordin RB, Lock HN. Chronic neuronopathic type of Gauchers disease with progressive myoclonic epilepsy in the absence of visceromegaly and bone involvement. Scott Med J. 2014; 59(2):e1-6. [ Links ]
105. Weinreb NJ, Charrow J, Andersson HC, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 2002; 113(2):112-9. [ Links ]
106. Blocklet D, Abramowicz M, Schoutens A. Bone, bone marrow, and MIBI scintigraphic findings in Gauchers disease «bone crisis». Clin Nucl Med. 2001; 26(9):765-9. [ Links ]
Contribución de autoría:
Los autores son integrantes del Grupo Peruano de Diagnóstico y Tratamiento de la Enfermedad de Gaucher.
Correspondencia:
Janetliz Cucho Jurado
Correo electrónico: janetlizcuchojcj@hotmail.com
Recibido: 03/12/2014
Aceptado: 21/05/2015

