










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Medica Herediana
versión impresa ISSN 1018-130X
Rev Med Hered vol.26 no.4 Lima oct. 2015
REPORTE DE CASO
Incontinentia pigmenti (Síndrome de Bloch-Sulzberger) en un paciente varón. Reporte de un caso
Incontinentia pigmenti (Bloch-Sulzberger Syndrome) in a male patient. A case report
Noé Atamari-Anahui 1,2,a , Sendy Solórzano-Gutiérrez 1,3,b
1 Facultad de Medicina Humana, Universidad Nacional de San Antonio Abad del Cusco. Cusco, Perú.
2 Asociación Científica de Estudiantes de Medicina Humana del Cusco (ASOCIEMH-CUSCO). Cusco, Perú.
3 Hospital Regional del Cusco. Cusco, Perú
a Estudiante de Medicina; b Médico dermatóloga
RESUMEN
Incontinentia pigmenti es una rara genodermatosis ligada al cromosoma X caracterizada por lesiones ampollares distribuidas sobre las líneas de Blaschko. Esta se presenta en cuatro estadios: vesicular, verrugoso, hiperpigmentado y atrófico. Es más frecuente en mujeres por su letalidad en varones, aunque hay casos de sobrevivencia en ellos. Se presenta el caso de un varón de 30 días de nacido que presentó lesiones vesiculo-ampollares de distribución lineal siguiendo las líneas de Blaschko. Se le realizó una biopsia cutánea cuya conclusión fue incontinentia pigmenti en estadio vesicular. Este es el primer caso varón reportado en la literatura peruana.
PALABRAS CLAVE: Incontinencia pigmentaria, enfermedades cutáneas genéticas, masculino. (Fuente: DeCS-BIREME).
SUMMARY
Incontinentia pigmenti is a rare X-linked dermatosis characterized by bullous lesions distributed along Blaschko´s lines. Four clinical stages are recognized: blister, verrucous or wart like lesions, hyperpigmentation and atrophic lesions. We present the case of a 30-day old male patient who presented with blisters and bullous lesions distributed along Blaschko´s lines. A skin biopsy was performed that confirmed the diagnosis. This is the first report of a male patient in Peru wth the syndrome.
KEYWORDS: Incontinentia pigmenti, Skin Diseases, Genetic, male. (Source: MeSH NLM).
INTRODUCCIÓN
Incontinentia pigmenti o síndrome de Bloch-Sulzerger es una alteración hereditaria dominante ligada al cromosoma X que afecta a múltiples sistemas y se caracteriza por lesiones ampollares en bandas, lineales o salpicadas que siguen las líneas de Blaschko y que suelen estar presentes o aparecen poco después del nacimiento (1).
Esta es consecuencia de una mutación en el modulador esencial del factor nuclear κB (NFκB) (NEMO) que ha sido localizado en el Xq28 (2). Se presenta en mayor frecuencia en mujeres debido a la mortalidad prenatal en varones, aunque se han descrito casos de sobrevivencia en varones con un genotipo XXY o mosaicismos somáticos (3). Se pueden diferenciar cuatro estadios: vesicular, verrugoso, hiperpigmentado y atrófico; donde los pacientes pueden no desarrollar todos los estadios o bien que varios estadios se superpongan (1).
Incontinentia pigmenti puede estar asociado a anormalidades sistémicas como alopecia, distrofia de las uñas, alteraciones en la dentición y ocasionalmente alteraciones de sistema nervioso central, alteraciones oculares o problemas estructurales del desarrollo (4-6).
El objetivo del reporte fue presentar un paciente masculino con esta enfermedad, sin complicaciones; se discute el cuadro clínico y se revisa la literatura.
PRESENTACIÓN DEL CASO
Varón de 30 días de nacido, de parto eutócico a término y con peso adecuado para la edad gestacional presentó desde el nacimiento lesiones vesiculo-ampollares distribuidas en la cara interna del miembro inferior (MI) derecho y la cara externa del miembro superior (MS) del mismo lado; siguiendo las líneas de Blaschko. Se le dejó en observación para ver si ocurría remisión espontánea.
Cuatro días después el paciente retornó al servicio presentando aumento del número de lesiones. El examen físico general no mostró alteraciones encontrándose dentro de límites normales. Al examen físico dermatológico se observaron múltiples lesiones vesiculo-ampollares, firmes, turbias, acompañadas de costras séricas y serohemáticas asentadas sobre una base eritematosa de distribución lineal que afectaban el borde interno del MI derecho y el borde externo del MS ipsilateral (Figura 1).
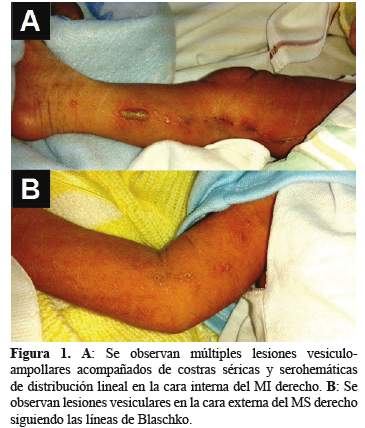
El hemograma mostró hemoglobina 15,3 g/dl; leucocitos 15 200 cél./mm3; neutrófilos 65%; linfocitos 22%; eosinófilos 10%; plaquetas 350 000 cél./mm3. Además, un examen directo y cultivo para hongos y bacterias resultó negativo. Se le realizó una biopsia cutánea de una de las vesículas del MI cuyo informe anátomo-patológico fue dermatitis perivascular superficial, espongiótica eosinofílica con presencia de vesículas intraepidérmicas que contienen numerosos eosinófilos, acantosis y espongiosis (Figura 2 (A)). Además, en la dermis se observó infiltrado inflamatorio de distribución perivascular superficial e intersticial conformado por linfocitos, eosinófilos y melanófogos (Figura 2(B)).
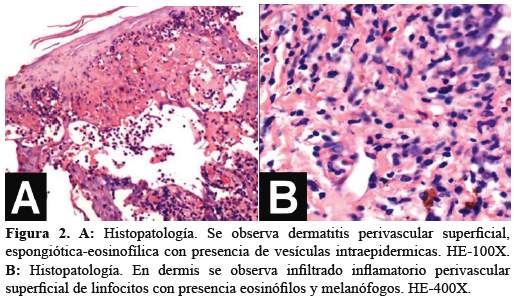
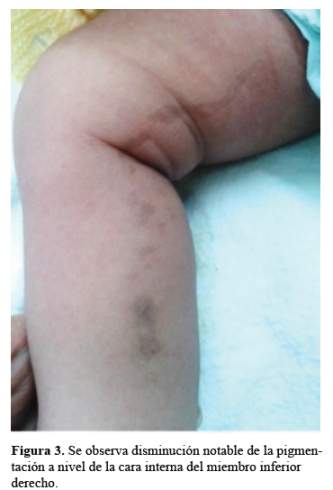
El diagnóstico anátomo-patológico final fue Incontinentia pigmenti en estadio vesicular. Se le realizó examen de fondo de ojo, no encontrando afección alguna. Se le indicó tratamiento con limpiadores e hidratantes de piel apreciándose en el control a los 30 días una disminución considerable de la pigmentación y evolución favorable (Figura 3).
DISCUSIÓN
El síndrome de Bloch-Sulzberger (Incontinentia pigmenti) es una rara genodermatosis ligada al cromosoma X. Fue descrito inicialmente por Garrod hacia el año 1906; sin embargo Bardach, Bloch, Siemens y Sulzberger definieron la condición posteriormente (7,8).
Afecta predominantemente a las mujeres a razón de 35:1 pues en los varones es de carácter letal por las complicaciones intrauterinas que se presentan. Su prevalencia en general es de alrededor de 1 por 40 000 (9).
Incontinentia pigmenti se caracteriza por lesiones cutáneas lineales que comienzan en el nacimiento y la evolución espontánea en cuatro estadios. El estadio 1 es inflamatorio vesiculo-ampolloso con bandas de vesículas epidérmicas llenas de eosinófilos que generalmente están presentes al nacer o en las primeras semanas de vida como lo evidenciado en el paciente y que se resuelve a los 4-6 meses de edad (1,8). En el estadio 2 se aprecian lesiones verrugosas, hiperqueratósicas que pueden aparecer en diferentes áreas distintas al estadio 1, estas se resuelven en 6 meses (8). El estadio 3 consiste en máculas con hiperpigmentación lineal que aumentan en forma gradual siguiendo las líneas Blaschko y un cuarto estadio de hipopigmentación y atrofia.
La anatomía patológica es importante pues nos ayuda al diagnóstico como en este caso en el que se evidenciaba vesículas intraepidérmicas con numerosos eosinófilos característico del estadio 1. Además de ello es importante el diagnóstico diferencial en cada estadio con otras entidades y de esta manera no pasar por alto la sospecha de Incontinentia pigmenti. El primer estadio puede confundirse con infecciones (bacterianas y por virus del herpes simple), eritema tóxico y epidermólisis ampollosa. El segundo estadio con el nevo epidérmico lineal; así como la hipermelanosis nevoide lineal y arremolinada que puede ser idéntica al tercer estadio.
Este caso nos muestra el desarrollo de la primera etapa en un paciente varón que no presentó complicaciones en el embarazo con desenlace fatal como describe la literatura. La supervivencia de los casos varones reportados tiene tres posibles mecanismos; presencia de cariotipos anormales; mutaciones hipomórficas y el mosaicismo (3). Desconocemos cuál de estos mecanismos presentó el paciente pues no se le realizó un estudio genético; aunque estudios demuestran que la explicación más probable de supervivencia en los pacientes masculinos es el resultado de mosaicismo somático por una mutación post-cigótica que se produce durante la etapa de blastocisto de la embriogénesis (10).
El diagnóstico oportuno y el seguimiento constante de estos pacientes es importante por las complicaciones sistémicas a largo plazo que pueden desarrollar. Este sería el primer caso de sexo masculino reportado en la literatura peruana el cual nos ayuda a comprender y sospechar esta patología (11-15).
REFERENCIAS BIBLIOGRÁFICAS
1. Morrell DS, Burkhart CN, Siegel D. Enfermedades hereditarias seleccionadas. In: Eichenfield L, Frieden I, Esterly N (eds.). Dermatologia Neonatal. 2da ed. Barcelona: Elsevier; 2009. p. 485. [ Links ]
2. Fusco F, Bardaro T, Fimiani G, et al. Molecular analysis of the genetic defect in a large cohort of IP patients and identification of novel NEMO mutations interfering with NF-kappaB activation. Hum Mol Genet. 2004;13(16):1763-73. [ Links ]
3. Kenwrick S, Woffendin H, Jakins T, et al. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter syndrome. Am J Hum Genet. 2001; 69(6):1210-7. [ Links ]
4. Pipa A, González M, López-Arranz E, Fernández J. Incontinencia Pigmentaria consideraciones odontoestomatológicas: Profilaxis y terapéutica. Av En Odontoestomatol. 2005;21(4):211-5. [ Links ]
5. Kataguiri P, Martins FCR, Yamada V, Salomão G, Ribeiro R, Rehder JRCL. Multiple clinical manifestations and diagnostic challenges of incontinentia pigmenti Syndrome. Rev Bras Oftalmol. 2010; 69(6):395-9. [ Links ]
6. Minic S, Trpinac D, Obradovic M. Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet J Rare Dis. 2013; 8:25. [ Links ]
7. Berlin AL, Paller AS, Chan LS. Incontinentia pigmenti: a review and update on the molecular basis of pathophysiology. J Am Acad Dermatol. 2002; 47(2):169-187. [ Links ]
8. Donnai D. Incontinentia pigmenti. In: Irvine AD, Hoeger PH, Yan AC (eds.). Harpers Textbook of Pediatric Dermatology. 3rd ed. Wiley-Blackwell; 2011. p. 1725-31. [ Links ]
9. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993; 30(1):53-9. [ Links ]
10. Pacheco TR, Levy M, Collyer JC, et al. Incontinentia pigmenti in male patients. J Am Acad Dermatol. 2006; 55(2):251-5. [ Links ]
11. Luy M, Noriega E, Llosa G. Incontinentia pigmenti: Sindrome de Bloch-Sulzberger Comunicación de un caso. Rev peru pediatr. 1963; 21: 144-49. [ Links ]
12. Arana G, Ricse H, Small O. Incontinentia pigmenti clásica. Tribuna Médica. 1976; 40(473): 30-2. [ Links ]
13. Tori C, Villar E, Arias J, Avalos C. Incontinentia pigmenti: A propósito de un caso. Rev Med Hered. 1995; 6(3):140-4. [ Links ]
14. Leyva-Sartori M, Cortez-Franco F, Carahyua-Pérez D. Incontinencia pigmenti: Reporte de un caso. Dermatol Perú. 2006; 16(1): 70-73. [ Links ]
15. Díaz E, Balbin E, Pereda J. Incontinencia pigmentaria (Síndrome Bloch-Sulzberger): Reporte de casos. Rev peru pediatr. 2012; 65(2): 94-100. [ Links ]
Agradecimiento:
A la Dra. María Esther Sanz por su colaboración en la identificación de la biopsia y toma de imágenes.
Declaración de financiamiento y de conflictos de intereses:
El reporte fue financiado por los investigadores quienes declaran no tener conflictos de intereses.
Contribución de autoría:
NAA y SSG participaron en la concepción, diseño del reporte. Todos los autores participaron en la recolección, análisis e interpretación de datos, así como en la revisión crítica del artículo y aprobaron su versión a publicar.
Correspondencia:
Noé Atamari-Anahui
Dirección: Calle los Geranios B-2, San Sebastián, Cusco, Perú
Teléfono: (+51) 958237215
Correo electrónico: noe.atamari@gmail.com
Recibido: 07/02/2015
Aceptado: 21/08/2015
