










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Medica Herediana
versión impresa ISSN 1018-130X
Rev Med Hered vol.26 no.4 Lima oct. 2015
TEMA DE REVISIÓN
Diagnóstico y tratamiento de la púrpura trombocitopénica inmunológica
Diagnosis and treatment of Immune thrombocytopenic purpura
Wilson Ruiz Gil 1,a;2,b
1 Facultad de Medicina Alberto Hurtado, Universidad Peruana Cayetano Heredia. Lima Perú.
2 Servicio de Hemato-Oncología, Hospital Nacional Cayetano Heredia, Ministerio de Salud. Lima, Perú.
a Profesor Principal de Medicina ; b Hematólogo.
RESUMEN
Desde siempre los episodios de sangrado muco-cutáneos, al ser tan evidentes, causan mucha preocupación y zozobra, más aún si ocurren sin causa aparente. La púrpura trombocitopénica suele ser la responsable de estos desagradables sucesos. Esta revisión tiene por objetivo actualizar los conocimientos acerca de la fisiopatología, el diagnóstico y el tratamiento de la púrpura trombocitopénica inmunológica (PTI), una patología hematológica que afecta tanto niños como a adultos y que se ve con relativa frecuencia en la actividad diaria de un hospital general.
PALABRAS CLAVE: Púrpura trombocitopénica, plaquetas, púrpura, autoanticuerpos. (Fuente: DeCS BIREME).
SUMMARY
Thrombocytopenic purpura is mostly responsible for episodes of muco-cutaneous bleeding. This review updates topics on the pathophysiology, diagnosis and treatment of immunologic thrombocytopenic purpura (IPT), an hematologic condition that afects both childern and adults, which is seen relatively frequent in daily practice in a general hospital.
KEYWORDS: Purpura thrombocytopenic, Blood platelets, autoantibodies. (Source: MeSH NLM).
INTRODUCCIÓN
Se entiende por púrpura trombocitopénica inmunológica, la condición clínica que se caracteriza por la disminución de plaquetas en la sangre periférica (SP), mediada por anticuerpos dirigidos contra antígenos plaquetarios que aceleran su destrucción periférica e incluso pueden inhibir la producción de las mismas. La manifestación clínica predominante es la hemorragia muco-cutánea, siendo la más característica la petequia.
La mayoría de los casos son considerados primarios denominado Púrpura Trombocitopénica Idiopática (PTI), mientras que otros son secundarios o coexisten con otras condiciones clínicas (PTI secundarios). Esto implica que la PTI es una condición clínico-patológica que involucra desórdenes heterogéneos que tienen en común la producción de auto-anticuerpos contra las plaquetas. Por la misma razón el diagnóstico de PTI resulta siendo de exclusión, dado que no existe una prueba diagnóstica específica.
Fisiología: Trombopoyesis (1,2,3)
La cuenta normal de plaquetas en sangre periférica (SP) varía entre 150 y 450 x 103/mm3. Se denomina trombocitopenia cuando la cuenta plaquetaria es menor de 150 000 / mm3; pero, hay que recordar que hasta 2,5% de la población normal puede tener valores inferiores a éste.
Las plaquetas son producidas en la médula ósea (MO) a partir de los megacariocitos que mediante un peculiar método de reproducción, la endomitosis, se convierten en células poliploides (hasta 64 N, promedio 16 N), muy grandes y con citoplasma amplio, el que se va fragmentando y liberando paulatinamente en los sinusoides de la MO. Finalmente, los fragmentos grandes son convertidos en plaquetas en el parénquima pulmonar. La producción normal de plaquetas se estima en 1x 1011 por día, está producción puede llegar a incrementarse hasta ocho veces en condiciones de demanda extrema.
La producción plaquetaria está regulada, principalmente, por la trombopoyetina (TPO) que es una citoquina (familia de la eritropoyetina y de la G-CSF) elaborada en el hígado, el riñón y los músculos a una tasa más o menos constante y que esávidamente capturada por su receptor (CD110) que es una proteína codificada por el gen c-Mpl. Este receptor, que posee dos dominios extracelulares y dos intracelulares, está presente en la superficie de las plaquetas y megacariocitos (adultos y jóvenes), pero también en los otros precursores hematopoyéticos. La caída marcada de las plaquetas deja mayor cantidad de TPO libre capaz de estimular a los megacariocitos, especialmente a los más jóvenes, permitiendo de esta manera la recuperación de las mismas.
Las plaquetas tienen un tiempo de vida media de 7 a 9 días; al envejecer son retiradas de la circulación por el sistema monocito macrófago, como resultado de una muerte programada (apoptosis). Hasta un tercio del total de plaquetas periféricas están reservadas en el bazo y se mantienen en equilibrio con el resto de plaquetas circulantes (4).
Desde un punto de vista fisiológico, más importante que el número total de plaquetas es la masa plaquetaria, que está en función no solo del número sino también del volumen plaquetario individual, que en condiciones normales varía entre 7 a 10 fl, pero en condiciones patológicas puede llegar a 30 fl o más (5).
Las plaquetas jóvenes contienen material RNA, similar a los reticulocitos de los glóbulos rojos (GR); esta característica permite que los nuevos contadores automatizados las puedan reconocer y cuantificar. La evaluación de estas Plaquetas reticuladas o fracción de plaquetas inmaduras (FPI), es una herramienta útil para medir la respuesta medular frente a las diferentes condiciones de trombocitopenia (6).
Hay que mencionar que una cuenta de plaquetas bajas puede corresponder a una pseudotrombocitopenia; se sabe que hasta el 0,1% de la población puede tener aglutininas contra las plaquetas, que se activan en presencia del EDTA y causan este fenómeno; esta anomalía se ve con más frecuencia con el uso de los equipos automatizados y de EDTA como anticoagulante. Por esta razón es una buena práctica el revisar la lámina de SP en simultáneo con la cuenta plaquetaria.
Fisiopatología de la trombocitopenia (7)
Clásicamente la púrpura trombocitopénica se ha clasificado en: a) Secundaria, aquella en la que se encuentra alguna patología asociada y; b) Primaria, a la que carece de dicha asociación; por lo tanto se llega al diagnóstico por descarte. La experiencia clínica muestra que 70 a 80% de los casos de púrpura trombocitopénica suele ser primaria y el otro 20 a 30% restante, secundaria.
Frente a una cuenta plaquetaria menor al mínimo normal se plantea la disyuntiva: ¿Hay un aumento en el consumo (o destrucción) o hay una falla en la producción?, en la tabla 1 se listan las probables causas.

La trombocitopenia debida a falla selectiva de la serie megacariocítica es muy inusual y podría corresponder a una rara entidad conocida como púrpura amegacariocítica adquirida (8), o se trataría de un estadio evolutivo en el curso de una anemia aplásica o de un síndrome mielodisplásico. Cuando el problema es el aumento en el consumo o destrucción periférica, lo más frecuente es que sea mediado por anticuerpos.
Trombocitopenia no inmune
Los mecanismos no inmunes a tener en cuenta son: La hemólisis microangiopática común en coagulación intravascular diseminada (CID), púrpura trombótica trombocitopénica (PTT), síndrome urémico hemolítico (SUH), síndrome HELP (anemia hemolítica, transaminasas elevadas y cuenta baja de plaquetas) en gestantes; hemangioma y metástasis carcinomatosa intravascular (9). Además, existe la acción directa de drogas tipo Ristocetina o alcohol.
Aparte de la ya mencionada pseudo trombocitopenia secundaria al uso de EDTA en donde la vida media de las plaquetas no está alterada; existen los casos del llamado secuestro esplénico, en donde el almacén esplénico de plaquetas puede incrementarse hasta el 90 % del total, como en la esplenomegalia por hipertensión portal y que rara vez se acompaña de sangrado clínico (4). Otra es la trombocitopenia por dilución como cuando un paciente recibe entre 15 a 20 paquetes de glóbulos rojos (PGR) en 24 horas. El problema se puede obviar usando un concentrado de plaquetas cada 10 a 15 PGR transfundidos.
Trombocitopenia mediada por anticuerpos
a) Trombocitopenia inmune secundaria (10)
Si bien no es el motivo principal de esta revisión, debemos mencionar que existe una serie de condiciones patológicas que pueden causar trombocitopenia mediada por anticuerpos como: Drogas (heparina, quinina); por anticuerpos alo-inmunes (post transfusión, post-trasplante, en periodo neonatal), por anticuerpos secundarios a otras patologías inmunes (Lupus, Síndrome antifosfolipídico, Artritis reumatoide), o a patologías neoplásicas (linfomas, LLC, cáncer de pulmón o de ovario); secundario a infecciones: tuberculosis (11), brucelosis (12), malaria, HIV, HCV, H. pilori (13).
b) Trombocitopenia inmune primaria (Autoinmune)
Harrington en 1951 demostró, en sí mismo, que la infusión de plasma de pacientes con PTI causaba una caída franca del número de plaquetas circulantes en el receptor. Luego, Shulman encontró que la causa de dicha trombocitopenia eran las inmunoglobulinas del tipo IgG. En 1982, van Leeuwen identificó a la glicoproteína IIb/IIIa, presente en la membrana plaquetaria, como el principal antígeno inductor de dichos auto-anticuerpos.
Muchos pacientes, especialmente aquellos con PTI crónico, tienen también anticuerpos contra otras glicoproteínas plaquetarias, como la Ib/IX (el receptor para el factor de von Willebrand) y Ia/IIa (un receptor de colágeno). Estas plaquetas marcadas con anticuerpos son luego secuestradas por el sistema monocito macrófago en el hígado, bazo y MO mediante sus receptores Fc-Re.
Actualmente se sabe que la tasa de destrucción plaquetaria es proporcional a la cantidad de anticuerpos fijos a su membrana (14).
Los avances en la comprensión de los mecanismos fisiológicos comprometidos con las reacciones inmunes tipo Th1 (citotóxica) y Th2 (humoral), han permitido entender mejor los procesos involucrados en la génesis del PTI y otros problemas de autoinmunidad.
Lo usual es que al diagnóstico el paciente ya tenga anticuerpos antiplaquetarios mono específicos (contra un solo antígeno) unidos a las plaquetas (Ab-plaq); el momento y la forma en que estos primeros anticuerpos aparecieron continua siendo un enigma (14,15).
Estos complejos Ab-plaq, son reconocidos por los macrófagos y monocitos (RES) mediante sus receptores Fc-Re, que luego de fagocitarlos y procesarlos generarán nuevos péptidos antigénicos, incluyendo glicoproteínas, lo cual conducirá al reclutamiento y activación de nuevas células T específicas que a su vez estimularán a nuevas células B a producir anticuerpos contra estos nuevos péptidos plaquetarios. Este fenómeno, conocido como ampliación de epítopes, sería el responsable de que la mayoría de los pacientes con PTI crónico de larga data, desarrollen anticuerpos contra múltiples glicoproteínas plaquetarias (14,15).
Entre otros mecanismos etiopatogénicos, se ha demostrado que las células T pueden producir directamente lisis de las plaquetas; así mismo se ha observado que los anticuerpos antiplaquetarios pueden ser tóxicos-inhibitorios a nivel de los megacariocitos, bloqueando la trombopoyesis, contribuyendo así a la trombocitopenia (14,16); este último mecanismo es el que ha dado origen a un nuevo enfoque terapéutico.
Del mismo modo que en otras condiciones de autoinmunidad en donde lo que ocurre en buena cuenta es una pérdida de la tolerancia inmunológica y un desbalance entre citoquinas, linfocitos B, T y macrófagos, en el PTI ocurriría lo mismo.
PRESENTACIÓN CLÍNICA (17)
La historia del sangrado sirve para orientar el diagnóstico. Cuando el problema es plaquetario, sea cuantitativo por ejemplo PTI o cualitativo ejemplo trombocitopatías, la hemorragia suele ser temprana, es decir inmediatamente después del trauma y de localización muco-cutánea, cuando es espontánea. Mientras que cuando se trata de un problema de factores de coagulación (humoral o tipo hemofilia) el sangrado post trauma suele ser tardío (varias horas después) y cuando es espontáneo predominan las hemorragias intra-cavitarias (hemartrosis) o viscerales (hematomas, tumores fantasmas). Esto se debe a que las plaquetas son las responsables del llamado trombo primario o inicial, mientras que los factores de la coagulación humoral se encargan de formar la malla de fibrina que, teniendo como base al trombo plaquetario y atrapando GR, termina formando el llamado trombo estable.
La intensidad y la frecuencia del sangrado guarda relación con el nivel de caída de las plaquetas, cuentas entre 150 hasta 30 x103 no suelen generar sangrado de manera espontánea, pero si con traumas. Mientras que recuentos plaquetarias menores de 30 x 103 presentarán hemorragia espontánea. Si las plaquetas bordean las 10 x 103/mm3 la posibilidad de complicaciones hemorrágicas severas se incrementa significativamente.
El signo más característico de la trombocitopenia es el sangrado que suele ser mucocutáneo. Cuando predominan las petequias y equimosis le suelen llamar Púrpura seca y cuando se acompaña (o predomina) de sangrado mucoso (epistaxis, gingival, menometrorragias) la llaman Púrpura húmeda.
La lesión cutánea por excelencia es la petequia que es una hemorragia en la piel a punto de partida de la micro-circulación (capilares o vénulas), rara vez mayor de 5 mm de diámetro, que suelen aparecer en las zonas de mayor presión (miembros inferiores), que no hacen relieve, que no son pruriginosas, que no desaparecen a la digito-presión y que evolucionan en varios días hasta desaparecer. También hay las llamadas equimosis que son hematomas superficiales de más de un centímetro y que no se diseminan más allá de la piel.
No suele haber crecimiento ganglionar ni vísceromegalia, salvo en población pediátrica en el que normalmente hasta en 10% se puede tener la punta del bazo palpable; pero, aún en estos casos, la presencia de esplenomegalia debe elevar fuertemente la sospecha de otro tipo de proceso.
El sangrado en áreas críticas, como el sistema nervioso central ocurre raras veces, generalmente post trauma, constituyéndose en la causa más común de muerte (de 2 a 4% de casos en gente mayor). Hay que mencionar que muchas veces los pacientes son asintomáticos y se descubre la trombocitopenia en un examen de rutina.
Siempre ha existido dificultad para evaluar los diferentes enfoques terapéuticos y ensayos clínicos en PTI por que los conceptos y niveles de trombocitopenia no han sido uniformes; recientemente un comité de expertos de la European Hematology Association (EHA), el Scientific Working Group on Thrombocytopenia (SWGT), ha propuesto un consenso para estandarizar la terminología y los criterios para definir la trombocitopenia clínicamente relevante, en niños y adultos (Tabla 2)(18).

De acuerdo a este consenso se considera trombocitopenia clínicamente relevante a cuentas de plaquetas menores a 100 x 103/mm3, de aquí en adelante seguiremos estos criterios.
PTI agudo del niño
Se presenta entre los 2 a 6 años de edad, no tiene predilección por sexo, es más frecuente durante los cambios de estación y usualmente hay la historia de un cuadro febril (viral) unos 7 a 21 días antes del inicio abrupto del cuadro hemorrágico. La incidencia es de 4,6 x 105 habitantes en Europa, mientras que en EEUU se reporta en 7,2 x 105.
El 90% de las veces la enfermedad se auto limita y desaparece en 3 a 6 meses. Si el cuadro perdura más tiempo se le considera persistente o crónico.
PTI crónico o del adulto
El inicio suele ser insidioso, episódico, en los adultos jóvenes tiene franca predilección por las mujeres (2 a 3:1 respecto al varón) no así en los mayores. La gran mayoría de casos son de curso crónico y más proclive a complicaciones y casi siempre van a requerir tratamiento.
En la literatura norteamericana se señala una prevalencia promedio de 23,6/100 000 personas, con predominio en las mujeres (28,1 vs 18,8 /105) y que se incrementa con la edad, 17,0/105 en el grupos de 18-49 años y de 36,2/105 en mayores de 65 años (19). Esto último demuestra que a diferencia de lo que se creía, el PTI es más prevalente en gente mayor.
EVALUACIÓN DIAGNÓSTICA
La evaluación diagnóstica se debe iniciar con la revisión de la lámina de SP para descartar las posibilidades de pseudotrombocitopenia o la satelización de los leucocitos por EDTA, o la presencia de megaplaquetas (anomalías congénitas), etc. Así mismo, en este examen se puede descartar la posibilidad de destrucción mecánica como ocurre en CID, PTT o SUH, que tienen en común la anemia hemolítica microangiopática.
Como no hay una prueba diagnóstica específica para el PTI se tiene que proceder por descarte. Para esto la historia y el cuadro clínico son los más importantes. Se deben solicitar las pruebas de laboratorio para eliminar las otras causas de trombocitopenia inmune como infecciones: virus HIV, hepatitis C (HCV), Brucella, TBC, o autoinmunes: lupus sistémico (LES) síndrome antifosfolipidico (SAF), artritis reumatoide (AR) y las neoplásicas como leucemia linfática crónica (LLC), linfoma, cáncer de pulmón, ovario, etc (Tabla 1).
Hay que mencionar que hasta 40% de los casos de PTI crónico del adulto pueden tener serología positiva para LES (20) o SAF (21) y hasta 13%, al cabo de algún tiempo, pueden evolucionar a LES (22). En cuanto a la trombocitopenia de la gestación sólo la evolución post parto puede diferenciarla del PTI clásico (23).
En el estudio de la MO se suele encontrar hipercelularidad a predominio de las series megacariocítica y en menor grado eritroide, la maduración muestra ligera desviación izquierda con predomino de estadios intermedios. En pacientes mayores de 50 años con sospecha de PTI este estudio sirve para el descarte, entre otros, de posibles Síndromes mielodisplásicos.
Muchos autores no recomiendan el estudio de la MO en casos pediátricos pero, sabiendo que el grupo etario en el cual aparece el PTI agudo del niño, es el mismo que el de la leucemia aguda y dado que la lectura de la SP no siempre se hace de manera correcta, soy de la opinión que en ciertos casos de PTI pediátrico se debe proceder a estudiar la MO.
La guía práctica para PTI de la Asociación Americana de Hematología (ASH) no recomienda el uso rutinario de las pruebas para detección de anticuerpos por su pobre sensibilidad y especificidad y porque el resultado no va a modificar el manejo del paciente (24,25) y tampoco sirven como predictores del tipo de evolución clínica (26).
TRATAMIENTO
PTI agudo
Como ya se mencionó entre 80 a 90% de los casos corresponde a niños y suele evolucionar espontáneamente, con una tasa de hemorragia intracraneana muy baja (<1%), por esto muchos especialistas proponen mantenerse en expectativa armada que consiste en no intervenir farmacológicamente y recomendar a los padres querestrinjan la actividad física del niño a lo mínimo necesario. Con frecuencia esta sugerencia no es aceptada por los padres.
La meta principal de todos los esquemas terapéuticos es lograr un nivel de plaquetas que asegure la ausencia de sangrado espontaneo, lo cual significa niveles mínimos de más o menos 50 000 /mm3.
El uso de corticoides suele ser el manejo inicial estándar, bajo modalidades diversas, que acortan el periodo de sangrado pero no garantizan que la enfermedad se termine de manera definitiva. Como siempre el uso prolongado de los esteroides acarrea los ya conocidos efectos colaterales.
En nuestra experiencia si el cuadro predominante es el de una púrpura húmeda o el sangrado cutáneo es muy florido o el niño es muy inquieto, se inicia prednisona (PRD) a dosis de 1 a 1,5 mg/kg/día hasta alcanzar una cuenta promedio de plaquetas de 100 000/mm3 lo cual ocurre en 10 – 20 días e iniciamos la disminución paulatina o tapering.
Si luego de esto, el cuadro hemorrágico se mantiene o la cuenta de plaquetas persiste alrededor de 30 000/mm3 se puede considerar: un nuevo curso de esteroide (es discutible) o usar otras alternativas como Inmunoglobulina G humana (IgG), Globulina anti D (Ig Anti D o Anti Rh), o tratar como PTI crónico.
De las inmunoglobulinas una de las más usadas es la Ig anti Rho (Anti D) básicamente en población pediátrica en razón de costos, pero hay que tener en cuenta que el paciente debe ser Rh (+) y no debe haber sido esplenectomizado ya que el modo de acción de esta terapia es inducir artificialmente un cuadro de anemia hemolítica aloinmune; se usa a dosis de: 50 a 75 μgr/kg IV.
La IgG humana es una buena alternativa cuando se necesita una respuesta más o menos rápida y segura. Se usa en dosis de 1g/kg/d IV por dos días; lo reservamos para casos de emergencia por razones de costo. Se debe recordar, en especial para los casos crónicos, que si bien brinda un alto porcentaje de respuestas (mayor al 90%), éstas son transitorias.
En edad pediátrica la opción de la esplenectomía se suele posponer hasta después de los 7 u 8 años de edad debido a la importancia que tiene este órgano en el proceso de maduración de las capacidades inmunológicas de los niños.
PTI crónico (Grafico 1)
Por definición el paciente que tenga cuentas plaquetarias menores a 100 000/mm3 por más de 12 meses se encuentra en esta categoría. Más del 80% de casos adultos y menos del 20% de los pediátricos corresponden a esta variedad.
Cuando la trombocitopenia dura entre 3 a 12 meses se denomina como PTI persistente (18).
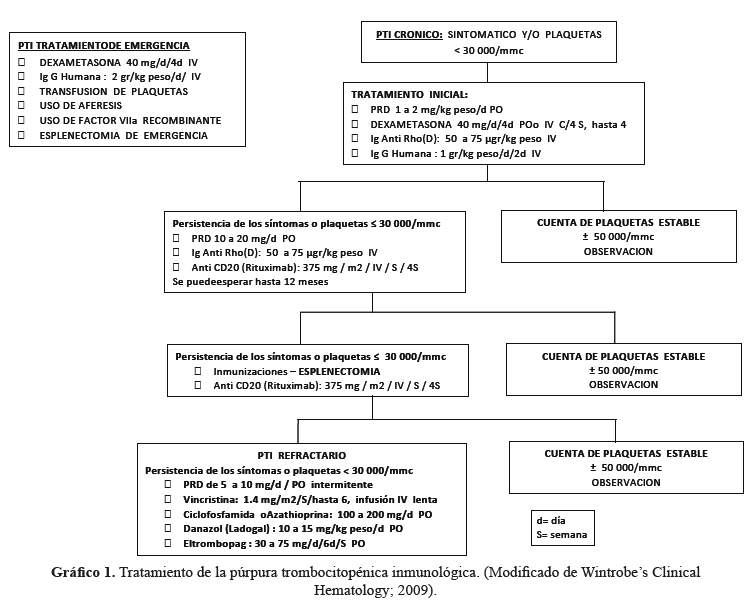
Si el paciente tiene más de 50 000/mm3 y no tiene sangrado, no va a exponerse a algún trauma quirúrgico en un futuro cercano o no ha respondido al uso estándar de esteroides (sobre todo en el caso pediátrico), la conducta debería ser expectante.
Los pacientes que se encuentren sintomáticos,3 es decir sangrando o persistan con plaquetas igual o menor de 30 000/mm3 deben iniciar tratamiento con corticoides, ya sea a dosis oral de 1 a 1,5 mg/kg/d o con alguno de los esquemas alternativos, como Dexametasona 40 mg/d/4d que puede ser oral o IV (27) y que se puede repetir mensualmente.
En los pacientes que no responden a cualquiera de estas alternativas se puede utilizar las opciones de segunda línea de tratamiento: la esplenectomía y en los últimos tiempos el uso de Rituximab y los nuevos agonistas del receptor de la trombopoyetina (TPO-agonistas). Estas nuevas herramientas han permitido replantear la táctica en el enfoque terapéutico del PTI crónico hasta en tres líneas (28). De los varios consensos de expertos publicados, el de la ASH me parece el más adecuado no solo por su peso institucional sino porque claramente sigue los principios de la Medicina basada en evidencias (Tabla 3) (29).

a) Esplenectomía
La esplenectomía es la opción terapéutica que claramente puede curar y la fuerza de la recomendación (1C) está avalada por más de 50 años de experiencia a nivel mundial. Los que no opinan a favor de esta opción lo hacen basándose en la posibilidad de una remisión espontanea, hecho que está reconocido pero que es muy poco frecuente, y en las conocidas (pero felizmente poco frecuentes) complicaciones infecciosas severas por gérmenes encapsulados que pueden ocurrir en pacientes esplenectomizados.
En la actualidad con la precaución de vacunar a los pacientes antes de la cirugía contra neumococo, Haemophilus y meningococo, el uso de nuevos antibióticos y la difusión de la cirugía laparoscópica, las complicaciones se han reducido mucho.
Con la esplenectomía los pacientes con PTI crónico sintomático o con plaquetas muy bajas, (≤30x103/mm3) tienen hasta 66% de posibilidades de curar (niveles normales de plaquetas y absolutamente asintomáticos) y hasta 22% de posibilidades de mejorar; es decir, asintomático pero con plaquetas menores a 100 000/ mm3 pero mayores de 50 000/mm3 (30).
De acuerdo a lo propuesto por el grupo de expertos de la ASH (29), el uso de Rituximab o de los TPOagonistas:
Eltrombopag y Romiplostim, como drogas de segunda línea, sólo deberían estar reservadas para aquellos casos en que no exista la posibilidad de realizar la esplenectomía.
b) Rituximab
Es un anticuerpo monoclonal quimérico (murino/humano) anti CD20 (Anti linfocito B) que inicialmente fue usado para tratar neoplasias de estirpe B (LLC, Linfomas NH) y luego ha demostrado eficacia en patologías en donde la participación de los linfocitos B es fundamental, entre otras condiciones los casos de PTI crónico.
Una revisión sistemática muestra una respuesta completa (plaquetas ≥150x103) en el 44% y respuestas parciales (plaquetas >50x103) en 19% con una duración menor a un año, en pacientes con PTI crónico (31).
Las complicaciones reportadas han sido escasas pero variadas: enfermedad del suero, leucoencefalopatia multifocal progresiva entre otros. Por estas razones el Comité de la ASH ha dado al Rituximab, una recomendación tipo 2B como tratamiento de segunda línea para el PTI crónico, mientras que para los casos de PTI crónico refractario la sugerencia es 2C.
c) Agonistas del receptor de la TPO (TPO-agonista) (32)
Cuando se comprendió que en la fisiopatología del PTI existía un componente de depresión en la producción plaquetaria y luego se pudo medir los niveles séricos de TPO en pacientes con PTI encontrándolos bajos en comparación con los niveles de TPO en pacientes con anemia aplásica, se abrió una nueva ventana terapéutica para drogas que estimularan la trombopoyesis. Cuando se pudo sintetizar la TPO se usó con fines terapéuticos pero tenía múltiples efectos colaterales.
Actualmente existen dos en el mercado:
El trombopag para uso oral y el Romiplostim para uso subcutáneo. Este último es un péptido que actúa en los dominios externos del receptor de la TPO, al igual que la trombopoyetina natural; fue el primero en ser usado en pacientes con PTI crónico que habían fracasado con las terapias convencionales. La dosis usual varía entre 1μgr /kg SC semanal, pudiendo llegar hasta 10 μgr /kg semanal.
Posteriormente ingresó el trombopag, con la ventaja de ser de uso oral. Se trata de un agonista de la TPO que no es un péptido y que actúa en los dominios de transmembrana del receptor, activando las vías JAK2 y STAT. La dosis recomendada fluctúa entre 25 mg/d hasta 75 mg/d.
Los efectos colaterales asociados al uso de estos TPO-agonista descritos son: el incremento en el riesgo de trombosis, la inducción de fibrosis medular, el riesgo de cataratas, alteraciones hepáticas, inducción de neoplasias y la trombocitopenia de rebote cuando se suspende la droga.
En la guía de la ASH para manejo del PTI crónico, los TPO agonistas merecen una potente recomendación 1B para su uso en tercera línea (PTI crónico refractario) y una recomendación 2C para su uso en segunda línea.
PTI crónico refractario
Son aquellos pacientes que luego de la esplenectomía no han alcanzado las metas mínimas, es decir continúan con sangrado o mantienen cuentas de plaquetas menores a 30 000/mm3. En estos casos se debería recurrir al Rituximab o a los TPO agonistas ya mencionados.
Debido a los costos muchas veces se opta por tratamientos tradicionales, que suelen ser más económicos y que sin llegar a ser espectacularmente efectivos, han contribuido a salvar vidas; entre ellos están: los alcaloides de la vinca, inmunosupresores (ciclofosfamida, azatioprina), PRD intermitente a dosis bajas, Ladogal (Danazol), ciclosporina A, micofenolato, etc.
En la experiencia de nuestro servicio, el uso de alcaloides de la vinca (Vincristina) en infusión intravenosa lenta (no menos de 6 horas) diluyendo 1 mg en 1000 cc de solución salina (cubierta de la luz) y repetida cada semana hasta un máximo de 5 o 6 dosis, suele dar buenos resultados con remisiones duraderas.
Tratamientos de emergencia Esta desesperada situación acontece cuando hay la sospecha o evidencia de sangrado que comprometa la vida del paciente, generalmente hemorragia intracraneal; en estos casos se justifica el uso de transfusión de plaquetas, esplenectomía de emergencia entre otras alternativas (Gráfico 1).
REFERENCIAS BIBLIOGRÁFICAS
1. Deutsch VR, Tomer A. Megakaryocyte development and platelet production. Br J Haematol. 2006; 134(5): 453. [ Links ]
2. Kaushansky K. Historical review: megakaryopoiesis and thrombopoiesis. Blood. 2008; 111(3): 981. [ Links ]
3. Amy E, Geddis M. Megakaryopoiesis. Semin Hematol. 2010; 47(3): 212–219. [ Links ]
4. Aster RH. Pooling of platelets in the spleen: role in the pathogenesis of hypersplenic thrombocytopenia. J Clin Invest. 1966; 45(5):645. [ Links ]
5. Thompson CB. From precursor to product, how do megakaryocytes produce platelets? In: Levin J, Williams N, Levine R. Megakaryocyte development and function. New York: Alan Liss Incorporated; 1986. p. 361. [ Links ]
6. Wang C, Smith B, Ault K, Rinder H. Reticulated platelets predict platelet count recovery following chemotherapy. Transfusion. 2002; 42(3): 368. [ Links ]
7. Rodger G. Thrombocytopenia: Pathophysiology and classification. In: Wintrobe M. Wintrobes clinical hematology. Philadelphia: Lippincott Williams & Wilkins. 2009. p. 1289. [ Links ]
8. Neeraj A, Joseph E, Spahr T, Werner D, Rodgers G. Acquired Amegakaryocytic Thrombocytopenic Purpura. Am J Hematol. 2006; 81:132–135. [ Links ]
9. Fishman A, Hoffman A, Volterra F, Frymus M, Gelluci M. Intracaval and intracardiac metastatic non seminomatous germ cell tumor: a rare cause of hemolytic anemia and thrombocytopenia. Cancer Invest. 2002; 20(7):996. [ Links ]
10. Douglas B, Liebman H, Stasi R. Pathobiology of secondary immune thrombocytopenia. Semin Hematol. 2009; 46(2): 2-14. [ Links ]
11. Madkaikar M, Ghosh K, Jijina F, Gupta M, Rajpurkar M, Mohanty D. Tuberculosis and immunethrombocytopenia. Haematologica. 2002; 87(8):38. [ Links ]
12. Sevinc A, Buyukberber N, Camci C, Buyukberber S, Karsligil T. Thrombocytopenia in Brucellosis: Case Report and Literature Review. J Natl Med Assoc. 2005; 97(2):290-293. [ Links ]
13. Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori associated chronic idiopathic thrombocytopenic purpura. Br J Haematol. 2004; 124:91–96. [ Links ]
14. Berchtold P, Wenger M. Autoantibodies against platelet glycoproteins in autoimmunes thrombocytopenic purpura: Their clinical significance and response to treatment. Blood. 1993; 81: 1246-1250. [ Links ]
15. Jhonsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology. 2012; 2012: 306-312. [ Links ]
16. Nugent D, McMillan R, Nichol JL, Slichter SJ. Pathogenesis of chronic immune thrombocytopenia: increased platelet destruction and/or decreased platelet production. Br J Haematol. 2009; 146(6):585. [ Links ]
17. Thinelt C, Calverley D. Thrombocytopenia caused by immunologic platelet destruction. In: Wintrobe M. Wintrobes clinical hematology. Philadelphia: Lippincott Williams & Wilkins;2009. p. 1292. [ Links ]
18. Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009; 113:2386-2393. [ Links ]
19. Feudjo-Tepie MA, Robinson N, Bennett D. Prevalence estimates of adult chronic Idiopathic Thrombocytopenic Púrpura (ITP) in the United States. ASH Annual Meeting Abstracts 2007; 110 (11): 3202. [ Links ]
20. Kurata Y, Miyagawa S, Kosugi S, et al. High-titerantinuclear antibodies, anti-SSA/Ro antibodies and anti-nuclear RNP antibodies in patients with idiopathic thrombocytopenic purpura. Thromb Haemost. 1994; 71(2): 184. [ Links ]
21. Bidot C, Jy W, Horstman L, et al. Antiphospholipid antibodies in immune thrombocytopenic púrpura tend to emerge in exacerbation and decline in remission. Br J Haematol. 2005; 128(3):366. [ Links ]
22. Mestanza-Peralta M, Ariza-Ariza R, Cardiel M, Alcocer-Varela J. Thrombocytopenic purpura as initial manifestation of systemic lupus erythematosus. J Rheumatol. 1997; 24(5):867. [ Links ]
23. Lescale K, Eddleman K, Cines D, et al. Antiplatelet antibody testing in thrombocytopenic pregnant women. Am J Obstet Gynecol. 1996; 174(3):1014. [ Links ]
24. George J, Woolf S, Raskob G, et al. Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood. 1996; 88(1):3. [ Links ]
25. Davoren A, Bussel J, Curtis B, Moghaddam M, Aster R, McFarland J. Prospective evaluation of a new platelet glycoprotein (GP)-specific assay (PakAuto) in the diagnosis of autoimmune thrombocytopenia (AITP). Am J Hematol. 2005; 78(3):193. [ Links ]
26. Taub J, Warrier I, Holtkamp C, et al. Characterization of autoantibodies against the platelets glycoprotein antigens IIb/IIIa in childhood idiopathic thrombocytopenia púrpura. Am J Hematol. 1995; 48:104-107. [ Links ]
27. Cheng Y, Wong R, Soo Y, et al. Initial treatment of immune thrombocytopenic purpura with high-dose dexamethasone. N Engl J Med. 2003; 349:831-836. [ Links ]
28. George J. Sequence of treatments for adults with primary immune thrombocytopenia. Am J Hematol. 2012; 87:S12–S15. [ Links ]
29. Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Crowther M. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011; 117(16): 4190-4207. [ Links ]
30. Kojouri K, Vesely S, Terrell D, George J. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: A systematic literature review to assess longterm platelet count responses, prediction of response, and surgical complications. Blood. 2004; 104:2623–2634. [ Links ]
31. Arnold D, Dentali F, Crowther M, et al. Systematic review: Efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med. 2007; 146:25–33. [ Links ]
32. Rice L. Treatment of immune thrombocytopenic purpura: focus on eltrombopag. Biologics. 2009; 3:151-157. [ Links ]
Correspondencia:
Dr. Wilson Ruiz Gil
Servicio de Hematología Oncología. Hospital Nacional Cayetano Heredia
Av Honorio Delgado No 262 Urb, Ingeniería. San Martin de Porras Lima 31, Perú.
Correo electrónico: wruizg@yahoo.es
Recibido: 06/06/2015
Aceptado: 13/07/2015
