










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Medica Herediana
versión impresa ISSN 1018-130X
Rev Med Hered vol.28 no.1 Lima ene. 2017
http://dx.doi.org/10.20453/rmh.v28i1.3077
CONVERSATORIO CLÍNICO
Caso clínico 01-2017. Mujer de 20 años con crecimiento de glándulas salivares y lacrimales, adenopatías, fiebre y lesión pulmonar
Clinical case 01-2017. A 20 year-old woman with lacrimal and salivary glands enlargement, lymphadenopathies, fever and a pulmonary lesion
Gran Ronda de Medicina Interna y Especialidades del Hospital Nacional Cayetano Heredia /Grand Round of Internal Medicine and Specialties at the Cayetano Heredia Hospital
Editor de sección: Dr. Sergio Vásquez Kunze.
Editores asociados: Dr. Héctor Sosa Valle, Dr. Leslie Soto Arquiñigo, Dra. Elena Zelaya Arteaga.
Sergio Vásquez 1,a, Fernando Mejía 2,a, Miro Rodriguez 3,a, Natalí Leiva 1,b, Kathy Gonzales 1,b
1 Servicio de Medicina Interna. Dpto. de Medicina, Hospital Cayetano Heredia. Lima, Perú.
2 Dpto. de Enfermedades Infecciosas, Tropicales y Dermatológicas, Hospital Cayetano Heredia. Lima, Perú.
3 Servicio de Oncología. Dpto. de Medicina, Hospital Cayetano Heredia. Lima, Perú.
a Médico Asistente; b Médico Residente de 3er año de Medicina Interna
PRESENTACIÓN DEL CASO
Natali Leiva Reyes (Residente 3er año Medicina Interna)
Mujer de 20 años, natural de Iquitos y procedente de Lima quien hace 4 semanas presentó aumento de volumen en el borde inferior del párpado superior izquierdo y una semana después del párpado superior derecho. Dos semanas antes del ingreso se agregó crecimiento parotídeo bilateral y en región submaxilar y una semana antes de la admisión se añadió otalgia bilateral, disfonía y xerostomía. Durante esa semana también notó aparición de un ganglio axilar derecho, no doloroso, sensación de alza térmica y tos no productiva por lo cual acudió al hospital, siendo hospitalizada. En las funciones biológicas tenía hiporexia, sed aumentada, sudoración normal y refería una pérdida de peso de 4 kg en el último mes. Las deposiciones y orina no mostraban alteraciones y estaba eutímica.
La paciente vivía en Lima hace varios años y era estudiante de administración. No ingería alcohol no fumaba ni usaba drogas. Su historia ginecológica era G: 0 P: 0, régimen catamenial 5/30, negaba relaciones sexuales y su FUR fue dos semanas antes del ingreso. No tenía antecedentes patológicos de importancia y previamente había estado sana. No había tenido intervenciones quirúrgicas. No tenía alergias a medicamentos. No tenía hermanos y sus padres no sufrían de enfermedades. En la revisión anamnésica de sistemas y aparatos negaba xeroftalmia, alopecia, artralgias o lesiones dérmicas.
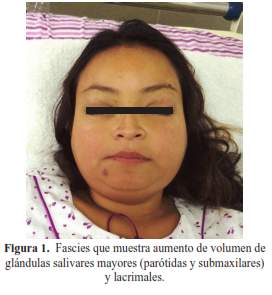
Al examen físico la PA: 100/60 mmHg FC: 78 x’ FR: 18 x’ T: 36,4°C; SAT: 97%; FiO2: 21%; peso: 54 kg; talla: 157 cm; IMC: 22,4. Lucía crónicamente enferma en regular estado general, hidratada, lúcida y orientada en las tres esferas, llamó la atención su fascies (figura 1). La piel, tejido celular subcutáneo y sistema osteomioarticular no mostraban alteraciones significativas. Se palpaba ganglio axilar derecho de 2 cm, no doloroso, móvil. Al examen regional se evidenció crecimiento de glándulas lacrimales, parótidas y glándula salivar submaxilar; el resto del cuello y las mamas, sin alteraciones. En tórax el murmullo vesicular pasaba bien en ambos campos pulmonares y no se auscultaron rales. En el examen cardiovascular, los ruidos cardiacos eran rítmicos, no habían soplos y los pulsos estaban conservados; no había ingurgitación yugular. El abdomen era plano, los ruidos hidroaéreos estaban presentes, y no se palpaban masas ni vísceromegalia. Al examen neurológico las pupilas eran reactivas a la luz y acomodación, fuerza muscular 5/5 simétrica, reflejos osteotendinosos 2/4, no había rigidez de nuca. El resto del examen era normal.
Los exámenes de laboratorio iniciales mostraronHb: 11,7 g/dl; hematocrito 34%; VMC: 86; HbMC:29,2; leucocitos: 5 780/mm3, plaquetas: 173 000/mm3, proteína C reactiva negativa. Glucosa 89 mg/dl; creatinina 0,5 mg/dl, DHL 1020 UI/l, electrolitos incluyendo calcio en valores normales; proteínas totales 6,8 g/dl, albumina 4 g/dl y resto del perfil hepático dentro de límites normales. El examen completo de orina fue normal. Las pruebas serológicas para VIH, Citomegalovirus, Epstein Barr virus y brucella fueron negativas. Se solicitaron factor reumatoideo, anticuerpos antinucleares (ANA), ANCA y perfil ENA (anti DNA nativo, anti Sm, anti RNP, anti Ro, anti La, anti Scl 70, anti histona y anti Jo1) que resultaron todos negativos. Los valores de subclases de IgG fueron: IgG1 770; IgG2 363; IgG3 33; IgG4 117, todas dentro del rango de referencia.
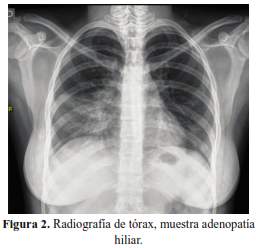
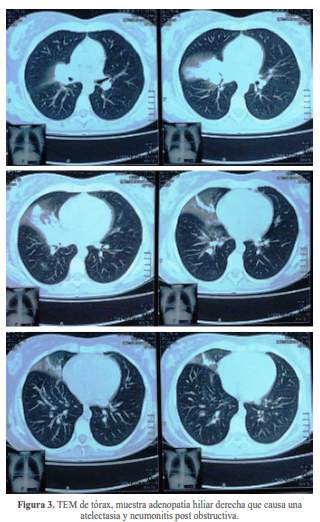
Se obtuvieron una radiografía de tórax (figura 2) y una tomografía (TEM) de tórax (figura 3). La TEM de abdomen fue no contributoria.
DISCUSIÓN
Dr. Sergio Vásquez Kunze (Medicina Interna)
Lo primero que hay que notar es que esta paciente tiene una fascies característica, aunque podría impresionar un edema facial y de cuello y confundirse con un síndrome de vena cava, el aumento de volumen es claramente dependiente de glándulas salivares mayores y lacrimales. La primera vez que vi esta fascies era residente de segundo año de Medicina y rotando en la unidad de cuidados intensivos; ingresó una paciente con artritis reumatoide con insuficiencia respiratoria, en ese momento se discutió que lo que tenía la paciente era un síndrome de Mickulicz, considerado como un síndrome de Sjogren severo secundario a su enfermedad de fondo. Al año siguiente vi un caso parecido, un paciente de reciente diagnóstico de VIH que ingresó con infiltración de glándulas salivares mayores y lacrimales, pero en ese caso el paciente tuvo diagnóstico de DILS (Síndrome de infiltración difusa de linfocitos), condición claramente asociada al VIH. Entonces, en primer lugar el síndrome de Mickulicz es un crecimiento de glándulas salivares mayores y menores con o sin afectación de glándulas lacrimales y corresponde a la presentación de la paciente, además nuestra paciente tiene fiebre y adenopatías, una de ellas de localización hiliar en la radiografía y TEM de tórax que causa una atelectasia y neumonitis post obstructiva. En segundo lugar, al ser este un síndrome tiene varias etiologías (tabla 1) que pasaré a discutir contrastando sus características con la enfermedad de la paciente.

Enfermedad de Sjogren
Enfermedad de presentación predominante en mujeres (9/1), sus manifestaciones cardinales son queratoconjuntivitis sicca (ojo seco) y xerostomía (boca seca). Puede tener también manifestaciones extra glandulares en casi todos los órganos en la mitad de los pacientes, siendo las adenopatías frecuentes. Puede ser una enfermedad primaria o secundaria, más comúnmente asociada a artritis reumatoide y lupus eritematoso sistémico. En el laboratorio los anticuerpos antinucleares son positivos en 90%, factor reumatoideo 70-90%, anti Ro 60%, anti La 40%. La xeroftalmia puede documentarse con un test de Schirmer (1). La biopsia de labio para glándulas salivares menores, muestra infiltración focal de linfocitos. Es importante saber que 5% de los pacientes pueden desarrollar linfoma, mayormente MALT y ocasionalmente grande difuso. Un marcador clínico de riesgo para esto es el agrandamiento parotídeo progresivo (2). Llamaba la atención que la paciente no tenía síntomas de xeroftalmia y que su serología de autoinmunidad era negativa. Esto, sumado a las adenopatías difusas y el marcado crecimiento parotídeo hacia el diagnóstico de Sjogren uno de exclusión.
Sarcoidosis
Su presentación clásica involucra lesiones pulmonares intersticiales, adenopatías (especialmente parahiliares), lesiones cutáneas y oculares. Puede involucrar glándulas salivares mayores principalmente parótidas y submaxilares, pero es infrecuente (3) La fiebre uvoparotidea de Heersfordt incluye crecimiento parotídeo prominente, uveítis, fiebre y compromiso de 7mo par craneal. La sarcoidosis es infrecuente en nuestro medio y nuestra paciente no tiene las lesiones intersticiales pulmonares típicas, si bien impresiona tener adenopatía hiliar derecha que ocasiona atelectasia y neumonitis post obstructiva, las adenopatías hiliares en sarcoidosis suelen ser simétricas y bilaterales.
Síndrome de Infiltración Difusa de Linfocitos (DILS)
Es una enfermedad exclusivamente asociada al VIH. Su frecuencia es baja, se presenta en alrededor e 5% de los pacientes con VIH. Se caracteriza por un agrandamiento de las glándulas salivares con o sin xerostomía persistente. Puede tener manifestaciones extra glandulares también. A diferencia del Sjogren no tiene serología positiva para ANA y demás anticuerpos y en la biopsia la infiltración de linfocitos es CD8 siendo en Sjogren CD4 (4,5). Este diagnóstico queda excluido en nuestra paciente por tener serología negativa para VIH.
Tuberculosis
La tuberculosis parotídea es rara y aún más rara, bilateral. Se puede asociar o no a tuberculosis pulmonar, la diseminación es por el conducto de Stenon o linfangitica (6). Es un diagnóstico muy improbable en nuestro paciente.
Linfoma
Linfoma como causa de síndrome de Mickulicz es muy raro y en la literatura hay escasos reportes de casos. Se ha reportado variedad MALT y linfoma no Hodgkin de células B grandes difuso (7,8). Sin embargo, esta posibilidad podría aumentar en nuestro paciente por las múltiples adenopatías que presenta, fiebre y baja de peso y debe descartarse con biopsias de ganglio obligatoriamente.
IgG4- RD (related disease)
En los años 90’ se reconoció un tipo de pancreatitis que producía dolor y colestasis con infiltración de células plasmáticas en el páncreas. Esta se denominó pancreatitis autoinmune. En el año 2000 se reconoció que esta condición estaba asociada a incremento de la IgG subclase 4 en sangre y también en la histología. Es lo que ahora se conoce como pancreatitis autoinmune tipo 1 que es una IgG4-RD (IgG4-panceratitis) (9). En el 2004, ya se había reconocido que el incremento de IgG4 se asocia a una gran variedad de enfermedades sistémicas que tienen como característica la infiltración y crecimiento de órganos debido a plasmocitos que marcan para IgG4 y muchas de estas también tienen IgG4 elevados en forma periférica, se les ha denominado IgG4 -RD (10,11). Así la infiltración de glándulas salivares y lacrimales cuya causa es solamente la infiltración por este tipo de plasmocitos se conoce ahora como IgG4-sialoadenitis y por su parecido a la primera descripción de Mickulicz en 1800 se le ha denominado Enfermedad de Mickulicz (recibiendo el nombre de enfermedad y no de síndrome cuando es debido a IgG4) (12-14).
Enfermedades clásicas como la tiroiditis de Riedel (15), la fibrosis retroperitoneal (enfermedad de Ormond), el pseudotumor orbitario (16) y de más reciente reconocimiento como la aortitis autoinmune o paquimeningitis idiopática (17) se conocen ahora que son enfermedades relacionadas a IgG4. En nuestro hospital el grupo de gastroenterología ha presentado hace poco un primer caso de pancreatitis autoinmune tipo 1 con valores elevado de IgG4 e infiltración de glándulas salivares y lacrimales. La enfermedad asociada a IgG4 puede tener también crecimiento de ganglios de forma difusa, incluyendo mediastinales e hiliares y que puede mimetizarse con linfoma o infecciones. Las biopsias pueden parecerse a un linfoma de bajo grado, pero cuando se les hace la tinción para IgG4 resulta positiva (18).
Esta enfermedad es una fuerte posibilidad para nuestro paciente pues tiene el fenotipo Mickulicz y también adenopatías además de síntomas constitucionales. También la haría de mejor pronóstico pues en general responden a corticosteroides. Sin embargo, los niveles de IgG4 están dentro de valores normales, aunque como se discutido pueden ser normales y tener los plasmocitos IgG4 en las biopsias de órganos afectados.
En conclusión, creo que los mejores diagnósticos para esta paciente son linfoma y enfermedad asociada a IgG4. El plan debe incluir biopsia de glándulas salivares empezando por labio y biopsia de una adenopatía, debiéndose realizarse inmunohistoquimica.
Anatomía patológica
Dra. Kathy Gonzales (Residente de 3er año de Medicina Interna)
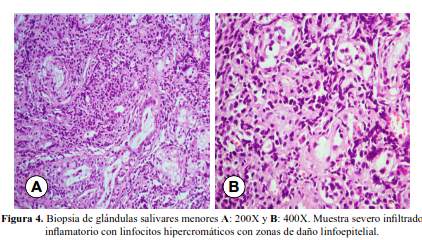
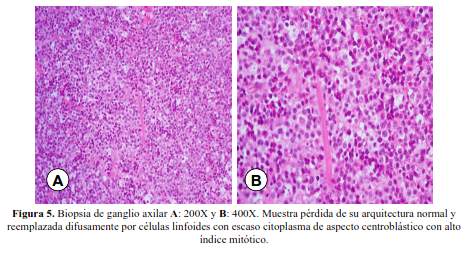
Se realizaron biopsias de glándulas salivares menores (figura 4) y del ganglio axilar (figura 5). El informe de la biopsia de glándulas salivares menores fue: “severo infiltrado inflamatorio con linfocitos hipercromáticos con zonas de daño linfoepitelial compatible con Síndrome de Sjogren-Mickulicz.” Se sugirió descartar linfoma de bajo grado con inmunohistoquimica. El informe de la biopsia del ganglio fue: “Ganglio linfático con pérdida de su arquitectura normal y reemplazado difusamente por células linfoides con escaso citoplasma de aspecto centroblástico con alto índice mitótico sugerente de linfoma no Hodgkin linfoblastico”. La inmunohistoquimica mostró CD3, CD4 y CD8 positivos, con Ki67 90% concluyente de un linfoma de células T maduras. Se realizó serología para HTLV 1, la cual fue positiva.
COMENTARIOS
Dr. Fernando Mejía Cordero (Enfermedades Infecciosas y Tropicales)
El virus linfotrópico a células T humanos tipo 1 (HTLV-1 por sus siglas en inglés), es el primer retrovirus descubierto. Se cree que veinte millones de individuos en todo el mundo están infectados con HTLV-1, la mayoría de ellos son asintomáticos. Los modos de transmisión son la lactancia materna, el contacto sexual, y la transfusión de componentes de células sanguíneas. El principal modo de transmisión en las zonas endémicas es a través de la lactancia materna como en el caso presentado. El estudio familiar del caso podría ayudar a definir la forma de transmisión y las medidas preventivas de transmisión en la familia.
El Perú es una zona endémica de infección por HTLV-1 con tasas de prevalencia entre 1-7% variando en diferentes regiones. Las enfermedades relacionadas con el HTLV-1 son diversas, siendo las complicaciones más graves la leucemia/linfoma de células T adultas (ATLL) o la mielopatía asociada al HTLV-1/paraparesia espástica tropical (PET). El riesgo de desarrollar estas condiciones se calcula entre el 1 y 5% para las enfermedades asociadas al HTLV-1 en general, incluyendo ATLL, PET, uveítis, polimiositis y artropatía, el riesgo de por vida puede ser cercano al 10%. Algunas infecciones graves como la estrongiloidiasis, la sarna costrosa o la dermatitis infectiva también se asocian a esta infección retroviral. La ATLL, es una de las manifestaciones más severas y de peor pronóstico. Existen varios tipos de ATLL: aguda, linfomatosa, crónica y smoldering o latente. Esta clasificación depende de las manifestaciones clínicas, la presencia de flower cells periféricas y órganos afectados. La ATLL de forma latente o crónica tiene un pronóstico relativamente bueno, Incluso sin tratamiento. Sin embargo, pueden evolucionar a ATLL aguda, que tiene un pobre pronóstico. Casi todos los pacientes con ATLL presentan linfadenopatía y el 50% tienen hepatoesplenomegalia. Las lesiones cutáneas también son comunes y pueden preceder o coincidir con la linfadenopatía o vísceromegalia.
La ATLL también puede afectar a los pulmones, el tracto gastrointestinal y el sistema nervioso central; la afectación de otros órganos es infrecuente. Llama la atención la forma y el tiempo de presentación del ATLL en esta paciente pues el tiempo de aparición es usualmente a partir de la tercera década de vida pues la infección retroviral requiere tiempo de latencia prolongado para desencadenar la neoplasia. Los pacientes con ATLL son inmunosuprimidos y tienen infecciones oportunistas, la neumonía por Pneumocystis jirovecii, meningitis criptocócica y herpes zoster son comunes en pacientes con ATLL.
La paciente presenta una forma tipo linfomatosa dada la afectación de órganos, la ausencia de flowers cells periférica y el compromiso sistémico. No existe tratamiento óptimo para la ATLL siendo la terapia antiretroviral adyuvante un factor de mejor supervivencia en los pacientes con formas latentes, crónicas y agudas, mas no en las linfomatosa como en este caso (19-21).
Dr. Miro Rodríguez Inocente (Oncología)
El cuadro clínico de la paciente orientaba definitivamente a Linfoma maligno, tenía un síndrome linfoproliferativo con presencia de síntomas constitucionales, DHL en más de 1000 UI/l, IgG4 normal, asociado a lesión linfoepitelial benigna, término con el que se conoce actualmente a la Enfermedad de Mickulicz que a su vez puede estar o no asociada a Síndrome de Sjogren. En ningún momento había un SOVCS ni clínico ni radiológico y lo que si llama la atención es que sea un Linfoma No Hodgkin a células T con serología positiva para HTLV-1 (transmisión materna), cuando los más frecuentemente asociados son los Linfomas de bajo de grado tipo MALT y células grandes difuso que son a células B, además de presentase este caso a una edad más temprana. El estudio Ki-67 en 90% habla de su alta agresividad, no tenemos el dato si la paciente tuvo o no estudio de médula ósea y biopsia de hueso para descarte de mieloptisis que probablemente lo tenga.
EVOLUCIÓN
Kathy Gonzales (Residente de 3er año de Medicina Interna)
La paciente inició quimioterapia para linfoma de células T con etoposido, prednisona, vincristina, ciclofosfamida y doxorubicina (EPOCH), a los pocos días desarrolló infiltración meníngea e hipercalcemia complicándose con neutropenia febril y choque séptico falleciendo a las 2 semanas del inicio de la quimioterapia.
DIAGNÓSTICO FINAL
- Linfoma de células T del adulto
- Infección por HTLV-1
- Síndrome de Mickulicz
REFERENCIAS BIBLIOGRÁFICAS
1. Shiboski SC, Shiboski CH, Criswell SA, et al. American College of Rheumatalogy classification criteria for Sjogren’s syndrome: A data-driven, expert consensus approach in the Sjogren’s International Collaborative Clinical Alliance Cohort. Arthritis Care Res. 2012; 64:45-87. [ Links ]
2. Fragkioudaki S1, Mavragani CP, Moutsopoulos HM.Predicting the risk for lymphoma development in Sjogren syndrome: An easy tool for clinical use. Medicine (Baltimore). 2016; 95(25):e3766.
3. Vairaktaris E, Vassilliou S, Yapijakis C, et al. Salivary gland manifestations of sarcoidosis: Report of three cases. J Oral Maxillofac Surg. 2005; 63:1016-21. [ Links ]
4. Itescu S, Brancato LJ, Buxbaum J, et al. A diffuse infiltrative CD8 lymphocytosis syndrome in human immunodeficiency virus (HIV) infection: a host immune response associated with HLA-DR5. Ann Intern Med. 1990; 112:3-10. [ Links ]
5. Ghrenassia E, Martis N, Boyer J, Burel-Vandenbos F, Mekinian A, Coppo P. The diffuse infiltrative lymphocytosis syndrome (DILS): A comprehensive review. J Autoimmun. 2015; 59:19-25. [ Links ]
6. Erkan AN, Cakmak O, Kayaselcuk F, Koksal F, Ozluoglu L. Bilateral parotid gland tuberculosis. Eur Arch Otorhinolaryngol. 2006; 263:487–9. [ Links ]
7. Revanappa MM1, Sattur AP, Naikmasur VG, Thakur AR.Disseminated non-Hodgkin’s lymphoma presenting as bilateral salivary gland enlargement: a case report. Imaging Sci Dent. 2013; 43(1):59-62.
8. Liu AC, Chen Y, Jia JS, Gao SY, Liu YY. Non-Hodgkin’s lymphoma mimicking Mikulicz disease: a case report. Beijing Da Xue Xue Bao. 2016; 48(6):1074-1076. [ Links ]
9. Zhao YX, Lü H. Autoimmune Pancreatitis: Typing, Diagnosis, and Treatment. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2016; 38(6):731-734. [ Links ]
10. Stone JH, Zen Y, Deshpande V: IgG4-related disease. N Engl J Med. 2012; 366:539-551. [ Links ]
11. Khosroshahi A, Stone JH: A clinical overview of IgG4-related systemic disease. Curr Opin Rheumatol. 2011; 23:57-66. [ Links ]
12. Yamamoto M, Takahashi H, Ohara M, et al. A new conceptualization for Mikulicz’s disease as an IgG4-related plasmacytic disease. Mod Rheumatol. 2006; 16:335-340. [ Links ]
13. Himi T, Takano K, Yamanoto M, et al. A novel concept of Mikulicz’s disease as Ig G4-related disease. Auris Nasus Larynx. 2012; 39: 9-17. [ Links ]
14. Harrison JD, Rodriquez-Justo M. Commentary on Ig G4-related sialadenitis: Mikulicz’s disease, Kittner tumour, and eponymy. Histopathology. 2011; 58:1164-66. [ Links ]
15. Dahlgren M, Khosroshahi A, Nielsen GP, et al. Riedel’s thyroiditis and multifocal fibrosclerosis are part of the IgG4-related systemic disease spectrum. Arthritis Care Res (Hoboken). 2010; 62:1312-1318. [ Links ]
16. Wallace ZS, Khosroshahi A, Jakobiec FA, et al. IgG4-related systemic disease as a cause of “idiopathic” orbital inflammation, including orbital myositis and trigeminal nerve involvement. Surv Ophthalmol. 2012; 57:26-33. [ Links ]
17. Riku S, Kato S. Idiopathic hypertrophic pachymeningitis. Neuropathology. 2003; 23:335-344. [ Links ]
18. Ihrler S, Harrison JD. Mikulicz’s disease and Mikulicz’s syndrome: analysis of the original case report of 1892 in the light of current knowledge identifies a MALT lymphoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005; 100:334-339. [ Links ]
19. Verdonck K, González E, VanDooren S, Vandamme AM, Vanham G, Gotuzzo E. Human T-lymphotropic virus 1: recent knowledge about an ancient infection.Lancet Infect Dis. 2007; 7(4):266–281. [ Links ]
20. Bazarbachi A, Suarez F, Fields P, Hermine O. How I treat adult T-cell leukemia/lymphoma. Blood. 2011; 118(7):1736–1745. [ Links ]
21. Matutes E. Adult T-cell leukaemia/lymphoma. Journal of Clinical Pathology. 2007; 60(12):1373-1377. [ Links ]
Recibido: 28/12/2016
