










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Medica Herediana
Print version ISSN 1018-130XOn-line version ISSN 1729-214X
Rev Med Hered vol.30 no.1 Lima Jan./Mar 2019
http://dx.doi.org/10.20453/rmh.v30i1.3471
REPORTE DE CASO
Síndrome de Frasier, primer caso reportado en Perú
Frasier´s syndrome: first reported case in Peru
Jenny Ponce Gambini 1, Reyner Loza Munarriz 1,2,a, Angélica Ynguil Muñoz 1, Sonia Chia Gonzales 3, Jaime Cok García 4
1 Departamento de Pediatría, Unidad de Nefrología pediátrica, Hospital Nacional Cayetano Heredia. Lima, Perú.
2 Facultad Medicina Alberto Hurtado, Universidad Peruana Cayetano Heredia. Lima, Perú.
3 Departamento de Medicina, Servicio de endocrinología, Hospital Cayetano Heredia. Lima Perú.
4 Departamento de Patología, Hospital Nacional Cayetano Heredia. Lima, Perú.
a Profesor Asociado.
RESUMEN
El síndrome de Frasier es una enfermedad rara producida por la mutación del gen WT1, caracterizado por pseudohermafroditismo masculino, disgenesia gonadal 46XY y enfermedad glomerular. Reportamos el caso de una mujer de 18 años, diagnosticada a los 12 años de síndrome nefrótico con desarrollo precoz de enfermedad renal crónica terminal y requerimiento de diálisis. A los 17 años se le detectó una tumoración abdominal. La laparotomía exploratoria reveló tumoración anexada a la trompa derecha que se extendía hasta la pared posterior del útero. Se le realizó histerectomía abdominal total con salpingo-ooforectomía bilateral. El diagnóstico anátomo-patológico fue disgerminoma. La paciente tenía amenorrea primaria y ausencia de caracteres sexuales secundarios. El estudio de corpúsculo de Barr de células de mucosa bucal fue negativo para cromatina sexual y el cariotipo fue 46, XY (Disgenesia Gonadal). El estudio genético reportó mutación heterocigótica en el intrón 9 del gen WT1. El cuadro es compatible con síndrome de Frasier, primer caso reportado en Perú.
PALABRAS CLAVE: Síndrome de Frasier, fallo renal crónico, disgerminoma.(Fuente: DeCS BIREME)
SUMMARY
Frasier´s syndrome is a rare disease caused by a mutation in the WT1 gene and is characterized by male pseudo hermaphroditism, gonadal dysgenesis 46XY and glomerular disease. We report the case of an 18-year-old female patient diagnosed at the age of 12 years of age of nephrotic syndrome who rapidly progressed to chronic failure needing dialysis. At the age of 17-years of age she presented an abdominal tumor, a laparotomy revealed a tumor attached to the right fallopian tube. An hysterectomy with bilateral fallopian tube extirpation, pathological findings revealed a dysgerminoma. The patient had a history of primary amenorrhea and absence of secondary sexual features. The study of Barr´s corpuscle of the oral mucosa was negative for sexual chromatin and the karyotype was 46 XY (gonadal dysgenesis). The genetic study revealed heterozygous mutation in the 9 gene WT1. The clinical presentation is compatible with Frasier´s syndrome, fist time reported in Peru.
KEYWORDS: Frasier syndrome, kidney failure, chronic, dysgerminoma. (Source: MeSH NLM).
INTRODUCCIÓN
El gen supresor de tumor de Wilms (WT1) juega un rol crítico durante el desarrollo del riñón y genitales, por lo que un defecto en este gen puede producir anormalidades en el sistema genitourinario, disfunción del podocito y predisposición al desarrollo de tumor de Wilms. Las mutaciones en este gen producen patologías sindrómicas, como Síndrome de WAGR, síndrome de Denys-Drash (SDD) y síndrome de Frasier (SF), así como casos aislados de tumor de Wilms y síndrome nefrótico (1).
Los pacientes con SF clásicamente presentan pseudohermafroditismo masculino (genitales externos femeninos normales, gónadas rudimentarias y cariotipo XY) y síndrome nefrótico de inicio en la niñez, debido usualmente a glomeruloesclerosis focal y segmentaria (GEFS), el cual progresa lentamente a enfermedad renal crónica terminal (ERCT) durante la segunda o tercera década de la vida (1), rara vez durante la niñez (2,3). Los pacientes con SF frecuentemente desarrollan gonadoblastoma e infrecuentemente tumor de Wilms (4), a diferencia del SDD quienes desarrollan con frecuencia tumor de Wilms (1,5,6).
El SF es causado por mutaciones en el intron 9 del gen WT1, siendo la mayoría mutaciones de novo (1,7). Sin embargo, se ha reportado que esta misma mutación puede producir GEFS con desarrollo genital adecuado en mujeres 46XX (8,9) y muy infrecuentemente, en varones 46XY (10). Por el contrario, la mutación en el exón 9 de este gen produce el SDD, caracterizado por la asociación de síndrome nefrótico de inicio temprano, debido a esclerosis mesangial difusa, con progresión rápida a ERCT, pseudohermafroditismo masculino y tumor de Wilms (1,6,11).
El SF ha sido considerada una enfermedad rara. Se han reportado algo más de 50 casos en la literatura (1), ninguno en Perú, por lo que el objetivo de este artículo fue presentar el primer caso de Síndrome de Frasier en nuestro país.
PRESENTACIÓN DEL CASO
Mujer de 18 años de edad, natural y procedente de la comunidad nativa de Poyeni, en Junín, Perú; cuarta hija de un matrimonio no consanguíneo. A los 12 años se diagnosticó síndrome nefrótico con deterioro rápido de la función renal, además de tuberculosis ganglionar. Se descartó amiloidosis mediante biopsia de glándulas salivales labiales. Ingresó al programa de diálisis peritoneal crónica ambulatoria (DPCA) y completó tratamiento específico para tuberculosis. A los 15 años ingresó al programa de hemodiálisis debido a peritonitis fúngica. A partir de los 17 años presentó episodios de dolor pélvico intermitente, retraso en el inicio de la pubertad y amenorrea primaria; al examen se encontró tumoración abdominal en el hipogastrio.
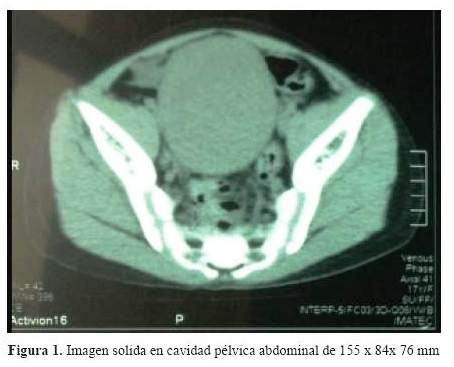
Se le realizó una TEM abdomino-pélvica la que mostró una imagen sólida de 155x84x76 mm, de morfología piriforme, localizado en mesogastrio e hipogastrio (figura 1). En la laparotomía exploratoria se encontró tumoración anexada a trompa derecha que se extendía hasta la pared posterior del útero de 3,5 cm, ovario izquierdo hipotrófico (1,4 x 0,9 x 0,4 cm) y trompas de Falopio normales. Se realizó histerectomía abdominal total con salpingo-ooforectomía bilateral.
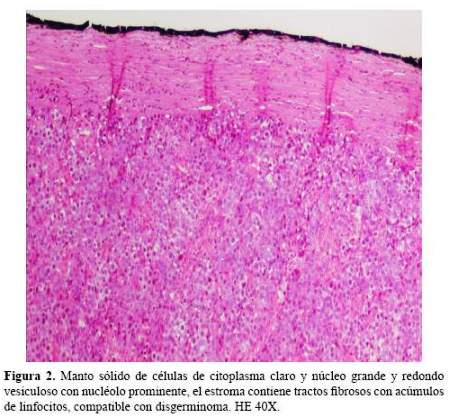
El estudio anátomo-patológico reveló disgerminoma estadio clínico IIIa (figura 2) y se inició quimioterapia con cis platino y etopósido (completó 3 ciclos). La gonadotropina coriónica humana se encontraba elevada y los otros marcadores tumorales, como CA 125, antígeno carcinoembrionario y alfa feto proteína, dentro de valores normales. FSH y LH estaban elevados y el estradiol disminuido, lo que indicó hipogonadismo hipergonadotrópico.
El estudio de corpúsculo de Barr fue negativo para cromatina sexual y el cariotipo fue 46XY. En el estudio genético se encontró mutación heterocigótica en el intrón 9 del gen WT1 (cortesía de Hildebrandt Laboratory, Boston Childrens Hospital, Division of Nephrology).
DISCUSIÓN
Se presenta el primer caso reportado en Perú de síndrome de Frasier, diagnóstico confirmado mediante la clínica y el estudio genético que reveló mutación en el intrón 9 del gen WT1. El SF es una enfermedad rara que fue descrita por primera vez por Frasier en 1964 (12). El diagnóstico se basó en la asociación de pseudohermafroditismo masculino como resultado de la disgenesia gonadal y síndrome nefrótico cortico- resistente que progresa lentamente a ERCT, en aproximadamente luego de 8 a 10 años del inicio de la proteinuria (1,2).
El mecanismo molecular por el cual la mutación del gen WT1 produce proteinuria no ha sido totalmente esclarecido. Li y colaboradores (7), encontraron que la mutación en el gen WT1 en SF produce una expresión anormal de las moléculas del podocito, importantes para la integridad de la barrera de filtración glomerular. Usualmente la proteinuria se detecta durante la niñez, entre los 2 a 6 años o incluso posteriormente como en el caso de nuestra paciente (1) y muy infrecuentemente antes del año de edad, reportándose un caso en una lactante de 6 meses (13). La proteinuria progresa a síndrome nefrótico cortico-resistente y sin respuesta a otro inmunosupresor, excepto en dos casos que tuvieron respuesta parcial al uso de ciclosporina (14).
La mayoría progresa a ERCT entre la segunda a tercera década de la vida (1,2). Nuestra paciente desarrolló ERCT a los 12 años de edad, similar al caso en China descrito por Chan (15) y los casos en Brasil reportados por Sanches Guaragna (16), cuyos pacientes desarrollaron ERCT entre la edad de 9 a 11 años. Solo se han reportado pocos casos de progresión a ERCT durante la niñez (2,3,13). El hallazgo histológico típico encontrado en la biopsia renal es GEFS (1,17). Sin embargo, se han reportado casos de SF que se presentan con esclerosis mesangial difusa (4). En nuestra paciente, no se realizó biopsia renal por que acudió al hospital en un estadio muy avanzado de la falla renal.
El SF típicamente está asociado con fenotipo femenino con ausencia de desarrollo puberal, genotipo masculino (cariotipo 46XY) y disgenesia gonadal que predispone al desarrollo de gonadoblastoma, tumor frecuentemente benigno, pero, en raros casos puede progresar a tumor maligno de células germinales, más frecuentemente disgerminoma, como el caso de nuestra paciente. También se han descrito otros tumores como teratoma inmaduro, tumor embrional y tumor del saco de York (13,18,19). Comparado con otras condiciones en el cual el pseudohermafroditismo masculino está presente, el riesgo de tumor gonadal es alto en SF (60-67%) (20). El riesgo de tumor gonadal también está presente en los casos de SF varones 46XY, pero no se ha reportado en SF mujer 46XX (20).
El SF usualmente se sospecha durante la evaluación de amenorrea primaria en pacientes con síndrome nefrótico o falla renal, algunas veces el diagnóstico se hace luego del trasplante renal (2,15). Los niveles plasmáticos de FSH y LH son altos y los niveles de estradiol bajos, lo que indica una amenorrea gonadal. El útero es pequeño e infantil y los trompas de Falopio, vagina y genitales externos son normales, lo que sugiere una acción defectuosa de la hormona anti-mulleriana (13); todos estos cambios fueron observados en nuestra paciente.
El SF se debe diagnosticar en forma temprana por el riesgo de desarrollo de tumor maligno de células germinales, lo cual puede prevenirse mediante la gonadectomía profiláctica bilateral. De ahí la importancia del conocimiento de esta patología tan poco frecuente en nuestro país.
REFERENCIAS BIBLIOGRÁFICAS
1. Niaudet P, Gubler MC. WT1 and glomerular diseases.Pediatr Nephrol. 2006; 21(11):1653-60. [ Links ]
2. Bache M, Dheu C, Doray B, et al. Frasier syndrome, a potential cause of end-stage renal failure in childhood. Pediatr Nephrol. 2010; 25(3):549-52. [ Links ]
3. Buzi F, Mella P, Pilotta A, Felappi B, Camerino G, Notarangelo LD. Frasier syndrome with childhood- onset renal failure. Horm Res. 2001; 55(2):77-80. [ Links ]
4. Barbosa AS, Hadjiathanasiou CG, Theodoridis C, et al. The same mutation affecting the splicing of WT1 gene is present on Frasier syndrome patients with or without Wilms tumor. Hum Mutat. 1999; 13(2):146-53. [ Links ]
5. Drash A, Sherman F, Hartmann WH, Blizzard RM. A syndrome of pseudohermaphroditism, Wilms tumor, hypertension, and degenerative renal disease. J Pediatr. 1970; 76(4):585-93. [ Links ]
6. Koziell A, Grundy R. Frasier and Denys-Drash syndromes: different disorders or part of a spectrum? Arch Dis Child. 1999; 81(4):365-9. [ Links ]
7. Li J, Zhao D, Ding J, et al. WT1 mutation and podocyte molecular expression in a Chinese Frasier syndrome patient. Pediatr Nephrol. 2007; 22(12):2133-6. [ Links ]
8. Peco-Antic A, Ozaltin F, Parezanovic V, Milosevski- Lomic G, Zdravkovic V. Proteinuria in Frasier syndrome. Srp Arh Celok Lek. 2013; 141(9-10):685-8. [ Links ]
9.Demmer L, Primack W, Loik V, Brown R, Therville N, McElreavey K. Frasier syndrome: a cause of focal segmental glomerulosclerosis in a 46XX female. J Am Soc Nephrol. 1999; 10(10):2215-8. [ Links ]
10. Tajima T, Sasaki S, Tanaka Y, et al. 46XY phenotypic male with focal segmental glomerulosclerosis caused by the WT1 splice site mutation. Horm Res. 2003;60(6): 302-5. [ Links ]
11.Little M, Wells C. A clinical overview of WT1 gene mutations. Hum Mutat. 1997; 9(3):209-25. [ Links ]
12.Frasier SD, Bashore RA, Mosier HD. Gonadoblastoma associated with pure gonadal dysgenesis in monozygous twins. J Pediatr. 1964; 64:740-5. [ Links ]
13. Gwin K, Cajaiba MM, Caminoa-Lizarralde A, Picazo ML, Nistal M, Reyes-Múgica M. Expanding the clinical spectrum of Frasier syndrome. Pediatr Dev Pathol Off J Soc Pediatr Pathol Paediatr Pathol Soc. 2008; 11(2):122-7. [ Links ]
14. Sinha A, Sharma S, Gulati A, et al. Frasier syndrome: early gonadoblastoma and cyclosporine responsiveness. Pediatr Nephrol. 2010; 25(10):2171-4. [ Links ]
15. Chan WKY, To KF, But WM, Lee KW. Frasier syndrome: a rare cause of delayed puberty. Hong Kong Med J. 2006; 12(3):225-7. [ Links ]
16. Guaragna MS, Lutaif ACG de B, Bittencourt VB, et al. Frasier syndrome: four new cases with unusual presentations. Arq Bras Endocrinol Metabol. 2012;56(8):525-32. [ Links ]
17. Gubler MC, Yang Y, Jeanpierre C, Barbaux S, Niaudet P. WT1, renal development, and glomerulopathies. Adv Nephrol Necker Hosp. 1999; 29:299-315. [ Links ]
18. Joki-Erkkila MM, Karikoski R, Rantala I. Lenko HL, Visakorpi T, Heinonen PK. Gonadoblastoma and dysgerminoma associated with XY gonadal dysgenesis in an adolescent with chronic renal failure: a case of Frasier syndrome. J Pediatr Adolesc Gynecol. 2002; 15(3): 145-9. [ Links ]
19. Shimoyama H, Nakajima M, Naka H, et al. A girl with bilateral ovarian tumours: Frasier syndrome. Eur J Pediatr. 2002; 161(2):81-3. [ Links ]
20. Ezaki J, Hashimoto K, Asano T, et al. Gonadal tumor in Frasier syndrome: a review and classification. Cancer Prev Res Phila Pa. 2015; 8(4):271-6. [ Links ]
Declaración de conflictos de interés: Los autores declaran no tener conflictos de interés.
Contribución de autoría: JPG, RLM, AYM, JCG: Descripción y discusión del caso; redacción del artículo.
Correspondencia:
Dr. Reyner Loza Munárriz
Correo electrónico: reyner.loza@upch.pe
Recibido: 25/09/2018
Aceptado: 29/12/2018
