










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Medica Herediana
versión impresa ISSN 1018-130Xversión On-line ISSN 1729-214X
Rev Med Hered vol.30 no.3 Lima jul. 2019
http://dx.doi.org/10.20453/rmh.v30i3.3585
REPORTE DE CASO
Hiperoxaluria primaria tipo 2 y oxalosis sistémica. Reporte de caso
Primary hyperoxaluria type 2 and systemic oxalosis. A case report
Reyner Loza Munarriz 1,2,a, Jenny Ponce Gambini 1, Angélica Ynguil Muñoz 1, Tatiana Sarmiento Barrientos 1, Jaime Cok García 3
1 Departamento de Pediatría, Servicio de Especialidades. Unidad de Nefrología pediátrica, Hospital Cayetano Heredia. Lima, Perú.
2 Departamento de Clínica Medicas, Sección de Pediatría, Facultad de Medicina Alberto Hurtado, Universidad Peruana Cayetano Heredia. Lima, Perú.
3 Departamento de Patología. Hospital Cayetano Heredia, Lima, Perú.
a Profesor Asociado.
RESUMEN
La hiperoxaluria primaria tipo 2 es una enfermedad rara caracterizada por sobreproducción de oxalato por una deficiencia enzimática intrahepática, lo que lleva a litiasis renal, nefrocalcinosis y daño renal crónico. Varón de 17 años con antecedentes de infecciones urinarias, y litiasis renal desde los 6 años. Desarrolló insuficiencia renal crónica terminal a los 11 años ingresando a terapia de diálisis peritoneal crónica ambulatoria. Durante su evolución presentó dolor a nivel de la columna vertebral y grandes articulaciones como tobillos, rodillas y hombros, deformación progresiva de las articulaciones distales de las manos. La resonancia magnética de columna reporto aplastamiento de cuerpos vertebrales en D8 y D9. La biopsia ósea de vértebra mostró depósito de cristales de oxalato de calcio. El estudio genético confirmó el diagnóstico de hiperoxaluria primaria tipo 2, esta enfermedad debe sospecharse en niños que forman cálculos a temprana edad, sería el primer caso reportado en Perú.
PALABRAS CLAVE: Hiperoxaluria, nefrocalcinosis, urolitiasis, insuficiencia renal crónica. (Fuente: DeCS BIREME)
SUMMARY
Primary hyperoxaluria type 2 is a rare disease characterized by over production of oxalate due to a deficiency of an intra hepatic enzyme leading to renal lithiasis, nephrocalcinosis and chronic kidney damage. We report the case of 17-year-old male patients with history of urinary tract infections and renal lithiasis since the age of 6 years. The patient developed end-stage kidney disease at the age of 11 years receiving chronic ambulatory peritoneal dialysis. He developed back pain and polyarthralgia of the ankles, knees, shoulders and progressive deformity of the hands. The magnetic resonance of the spine revealed flattening of D9-D9. Bone biopsy of the affected area showed presence of calcium oxalate. A genetic study confirmed the diagnosis of primary hyperoxaluria type 2. This entity should be suspected in children with renal stones at an early age, this may be the first case in Peru.
KEYWORDS: Hyperoxaluria, nephrocalcinosis, urolithiasis, renal insufficiency, chronic. (Source: MesH BIREME).
INTRODUCCIÓN
La hiperoxaluria primaria (HOP) es un error innato del metabolismo, con patrón de herencia autosómico recesivo, que resulta en una producción endógena incrementada de oxalato. Existen 3 tipos, siendo la más frecuente y más severa la HOP tipo 1 (1-3).
La HOP tipo 2 es causada por la deficiencia de una enzima citosólica con actividad de glioxilato reductasa, hidroxi-piruvato reductasa y D-glicerato deshidrogenasa (GRHPR) (4). Esta enzima tiene una distribución tisular amplia, primariamente intrahepática y su función es remover el glioxilato citosólico, mediante la reducción de glioxilato a glicolato e hidroxi-piruvato a D-glicerato, previniendo de esta manera su conversión a oxalato. En la deficiencia de esta enzima, la enzima lactato deshidrogenasa metaboliza el glioxilato acumulado a oxalato y la hidroxi-piruvato a L-glicerato (1,3). Por lo tanto, esta enfermedad se caracteriza por excreción urinaria elevada de oxalato de calcio y ácido L-glicérido, lo que ocasiona urolitiasis recurrente, nefrocalcinosis, enfermedad renal crónica progresiva y posteriormente depósito generalizado de oxalato de calcio en los diferentes tejidos (1-4). Típicamente, la HOP tipo 2 tiene un curso clínico menos severo y mejor pronóstico que la de tipo 1 (4,5). La nefrocalcinosis ocurre en el 12% de los pacientes, la afectación sistémica es rara y la progresión a enfermedad renal terminal (ERCT) es infrecuente. Esta última hasta ahora no se ha reportado en la edad pediátrica (6,7).
La incidencia de la HOP tipo 2 no es muy clara, ya que es una enfermedad extremadamente rara y muchos casos son reconocidos tardíamente o casi nunca son diagnosticados. Se han reportado menos de 30 casos en el mundo (5,6) y se han identificado 37 mutaciones en el gen GRHPR (1).
El objetivo del reporte fue presentar un caso de hiperoxaluria primaria tipo 2 de presentación inusual, con desenlace a ERCt y oxalosis sistémica, siendo éste es el primer caso reportado en nuestro país.
PRESENTACIÓN DEL CASO
Varón de 17 años, procedente de Piura, de padres consanguíneos, con historia de infecciones urinarias a repetición, nefrocalcinosis y eliminación de cálculos por la orina desde los 6 años. Se le diagnosticó ERCt a los 11 años y se inició diálisis peritoneal crónica ambulatoria. Durante el seguimiento, presentó dolores a nivel de columna y grandes articulaciones como tobillos, rodillas y hombros, deformación progresiva de las articulaciones distales de las manos y fracturas espontáneas, lo cual limitaba sus actividades diarias.
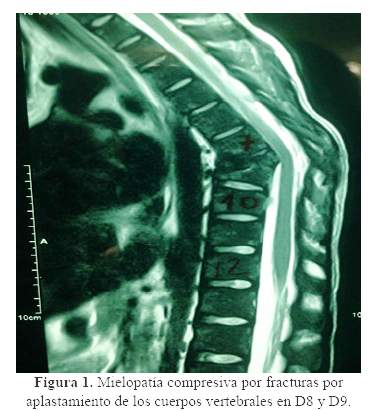
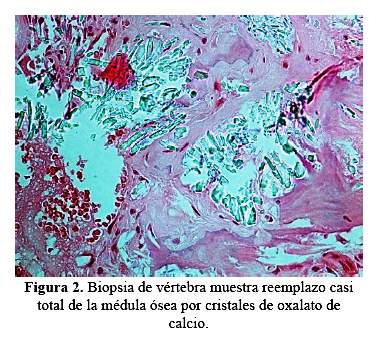
La resonancia magnética de columna mostró aplastamiento de cuerpos vertebrales en D8 y D9 (figura 1); la radiografía de abdomen mostró nefrocalcinosis y litiasis renal, y la uretro-cistografía retrógrada miccional, reflujo vésico-ureteral bilateral. En la biopsia ósea a nivel de vértebra se encontró extenso depósito de cristales de oxalato de calcio, dispuestos en forma radiada (figura 2). El estudio genético reportó mutación en hemicigosis en el gen GRHPR: c.295C>Tp. R99x (Mayo Clinic Hyperoxaluria Center).
El paciente inició hemodiálisis (HD) 3 veces/ semana, con filtro de alto flujo, concomitante con diálisis peritoneal automatizada y posteriormente, por falla de la membrana peritoneal, continuó sólo con HD 4 veces/semana, 4 horas al día, consiguiendo niveles de oxalato sérico dentro de valores normales (ácido oxálico:19,4 µmol/l, rango normal: 10-65 µmol/l).
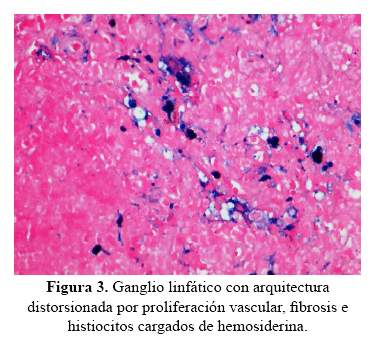
Durante su estancia en HD, presentó ascitis que requirió paracentesis evacuatoria en repetidas oportunidades. El estudio de ultrasonido mostró signos de hepatopatía crónica y la ultrasonografía Doppler y el estudio de líquido ascítico, signos de hipertensión portal. En la biopsia de hígado y ganglios se encontró depósitos de hemosiderina, diagnosticándose hemosiderosis hepática (figura 3).
Por el estudio genético, la clínica y la evolución se confirmó el diagnóstico de HOP.
DISCUSIÓN
La hiperoxaluria primaria puede ocurrir a cualquier edad, reportándose casos desde la infancia hasta la sexta década de la vida. La edad media de inicio es 5,5 años en la HOP tipo 1 (8,9) y 15 años en la HOP tipo 2 (7). La presentación clínica es heterogénea, varía desde casos severos que se presentan con nefrocalcinosis, deterioro progresivo de la función renal y oxalosis sistémica, a casos leves que se manifiesta únicamente con formación esporádica de cálculos en la edad adulta, incluso en algunos casos, el diagnóstico se hace por la recurrencia luego del trasplante renal (1,3,9).
El 20 a 50% de los pacientes con HOP tipo 1 e infrecuentemente con HOP tipo 2 progresan a ERCt (1,3). En la HOP tipo 1, la ERCt puede ocurrir en cualquier momento, entre los primeros meses y la sexta década de la vida, el 50% antes de los 25 años (9). No existen datos descritos en HOP tipo 2 en edad pediátrica ya que este desenlace es infrecuente. El diagnóstico de ERCt en nuestro paciente fue a los 11 años de edad, y sería el primer caso reportado de progresión a ERCt en la edad pediátrica.
Debido a la rareza de la enfermedad y al poco conocimiento de esta patología en la mayoría de países, se ha reportado un intervalo entre el inicio de los síntomas y el diagnóstico de 3 a 5 años, solo el 30% se diagnostica en forma precoz y 30% cuando ya tienen ERCt (8-10). En este caso, el diagnóstico se hizo en forma muy tardía, incluso varios años luego del diagnóstico de ERCt, y se sospechó por el hallazgo de cristales de oxalato de calcio en hueso y el antecedente de litiasis renal y nefrocalcinosis. Por la presencia de oxalosis sistémica se planteó inicialmente el diagnóstico de HOP tipo 1; sin embargo, el estudio genético reveló una HOP tipo 2, la cual como se manifestó párrafos arriba, lleva en forma infrecuente a ERCt y a oxalosis sistémica. En USA, de 90 pacientes diagnosticados con HOP hasta 1998 (79 con HOP tipo 1 y 9 con HOP tipo 2), ninguno de los pacientes con HOP tipo 2 tuvieron oxalosis sistémica y solo uno tenía ERCT al momento del diagnóstico (10).
El principal defecto en la hiperoxaluria es la sobreproducción de oxalato, principalmente en el hígado, por la deficiencia de la enzima GRHPR, lo que provoca aumento de la excreción de oxalato por el riñón, y condiciona a urolitiasis, nefrocalcinosis y afectación renal progresiva (1-3,7,9). El daño renal es causado por varios factores que en su conjunto producen disminución de la tasa de filtrado glomerular (TFG), estos incluyen a la toxicidad tubular por el oxalato, la inflamación producida por el depósito de oxalato de calcio intratubular y a nivel intersticial en la nefrocalcinosis y, la obstrucción renal debido a los cálculos (2).
Cuando la TFG es menos de 30-45 ml/min/1,73 m2 de superficie corporal, los riñones son incapaces de excretar efectivamente el exceso de oxalato; por ello, los niveles del mismo se elevan y superan su saturación, lo que produce que el oxalato se deposite en todos los tejidos (oxalosis sistémica), siendo los principales sitios de depósito los riñones y el hueso. Otros sitios de depósito son las paredes de los vasos sanguíneos, las articulaciones, la retina, la piel, médula ósea, corazón y el sistema nervioso central, lo que conduce a enfermedad grave e incluso la muerte (1–3,6,7). En nuestro paciente, otro factor adicional como causa de daño renal progresivo es el reflujo vésico-ureteral bilateral y los episodios frecuentes de infecciones del tracto urinario, lo que lleva a nefropatía por reflujo y formación de cicatrices renales. Esto, sumado al daño producido por el depósito de oxalato en riñón, descrito párrafos arriba, pudo haber condicionado a que una enfermedad que de por si rara vez lleva a daño renal crónico haya progresado a ERCt, oxalosis sistémica y daño óseo marcado.
En adición a la osteodistrofia renal relacionado directamente con la ERCt, los pacientes con HOP pueden experimentar dolor óseo severo, deformaciones óseas, fracturas óseas patológicas y anemia resistente a eritropoyetina y en casos más severos tofos subperiósticos (11). La biopsia ósea en estos pacientes muestra depósito de cristales de oxalato rodeados de reacción granulomatosa debido a la invasión de la superficie ósea por macrófagos (12). En nuestro caso, se encontró reemplazo casi total de la médula ósea por cristales de oxalato de calcio en la biopsia realizada a nivel de vértebra (figura 2), lo que explica la afectación ósea marcada en nuestro paciente y con gran impacto en la columna vertebral, que condicionó el aplastamiento vertebral (figura 1).
Asimismo, debido a que los dientes son tejidos mineralizados, nuestro paciente ha perdido la mayor parte de sus dientes debido al daño dental secundario al depósito de cristales de oxalato, similar al descrito en la biopsia ósea. En la literatura se han descrito menos de 12 casos con afectación de la cavidad oral en HOP. Los hallazgos consisten en resorción a nivel de mandíbula, resorción agresiva de la raíz externa, movilidad dental rápidamente progresiva, dolor dental, depósitos en el periodonto y pérdida ósea alveolar (11).
La presencia de hemosiderosis no está descrita en asociación con HOP, por lo que podríamos afirmar que se debe al suplemento de hierro administrado durante su terapia.
El diagnóstico definitivo de HOP en un paciente con signos y síntomas clínicos requiere pruebas genéticas para confirmar y determinar la variante mutacional, pues en forma infrecuente las manifestaciones clínicas en la HOP tipo 2 pueden ser igualmente severas que en la HOP tipo 1, como sucedió con nuestro paciente, y por lo que el tratamiento definitivo sería el trasplante combinado hepato-renal.
En la literatura, se han descrito aproximadamente 37 mutaciones en el gen GRHPR y la más común es la mutación c.103 del G en el exon 2, que representa el 40% de los casos (1,13). La mutación encontrada en nuestro paciente es poco frecuente. Se requieren más estudios que correlacionen el tipo de mutación con la clínica desarrollada.
Este reporte debe servir para llamar la atención sobre una enfermedad hasta ahora desconocida en nuestro país, por lo que se debe realizar estudios epidemiológicos y de screening en pacientes con litiasis renal recurrente y nefrocalcinosis, para detectar en forma precoz este tipo de patología de tan pobre pronóstico y así evitar la progresión a daño renal crónico.
REFERENCIAS BIBLIOGRÁFICAS
1. Rumsby G, Cochat P. Primary hyperoxaluria. N Engl J Med. 2013; 369(22):2163. [ Links ]
2. Harambat J, Fargue S, Bacchetta J, Acquaviva C, Cochat P. Primary hyperoxaluria. Int J Nephrol. 2011; 2011:864580. doi: 10.4061/2011/864580 [ Links ]
3. Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int. 2009; 75(12):1264-1271. [ Links ]
4. Johnson SA, Rumsby G, Cregeen D, Hulton S. Primary hyperoxaluria type 2 in children. Pediatr Nephrol Berl Ger. 2002; 17(8):597-601. [ Links ]
5. Milliner DS, Wilson DM, Smith LH. Phenotypic expression of primary hyperoxaluria: comparative features of types I and II. Kidney Int. 2001; 59(1):31-6. [ Links ]
6. Kemper MJ, Conrad S, Müller-Wiefel DE. Primary hyperoxaluria type 2. Eur J Pediatr. 1997; 156(7):509-512. [ Links ]
7. Leumann E, Hoppe B. The primary hyperoxalurias. J Am Soc Nephrol. 2001; 12(9):1986-1993. [ Links ]
8. Lieske JC, Monico CG, Holmes WS, et al. International registry for primary hyperoxaluria. Am J Nephrol. 2005; 25(3):290-296. [ Links ]
9. Cochat P, Liutkus A, Fargue S, Basmaison O, Ranchin B, Rolland MO. Primary hyperoxaluria type1: still challenging! Pediatr Nephrol 2006; 21(8):1075-1081. [ Links ]
10. Hoppe B, Langman CB. A United States survey on diagnosis, treatment, and outcome of primary hyperoxaluria. Pediatr Nephrol Berl Ger. 2003;18(10):986-991. [ Links ]
11. Bacchetta J, Boivin G, Cochat P. Bone impairment in primary hyperoxaluria: a review. Pediatr Nephrol Berl Ger. 2016; 31(1):1-6. [ Links ]
12. Bacchetta J, Farlay D, Abelin-Genevois K, Lebourg L, Cochat P, Boivin G. Bone impairment in oxalosis: An ultrastructural bone analysis. Bone. 2015; 81:161-167. [ Links ]
13. Takayama T, Takaoka N, Nagata M, et al. Ethnic differences in GRHPR mutations in patients with primary hyperoxaluria type 2. Clin Genet. 2014;86(4):342-348. [ Links ]
Declaración de financiamiento y de conflictos de intereses: Los autores declaran no tener conflictos de intereses.
Contribución de autoría: RLM, AYM, JPG, JCG, RMN, LZM: Descripción y discusión del caso; redacción del artículo.
Correspondencia:
Reyner Loza Munarriz
Correo electrónico: reyner.loza@upch.pe
Recibido: 04/04/2018
Aceptado: 28/06/2019
