











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El sarcoma de Ewing (ES) es un tumor óseo de huesos largos altamente maligno que se presenta en niños y adultos jóvenes; se han reportado casos de tumores malignos de tejido blando que son indistinguibles de ES y son llamados ES extra esqueléticos (EES); estos tumores son ahora clasificados como familia de tumores ES (ESFT); incluyen ES, EES y tumor neuro ectodérmico primitivo (PNET), que muestra más diferenciación neuronal que ES 1. EES es un tumor de partes blandas raro, agresivo y maligno con alta tasa de recurrencia y que ocurre principalmente en adolescentes y adultos jóvenes entre 10 y 30 años de edad 2. En 1975, Angervall y Enzinger informaron el primer caso de ES extraóseo 3.
ES y PNET forman un solo grupo de tumores del hueso y tejido blando; ES típicamente indiferenciado en un extremo del espectro y PNET con clara evidencia de diferenciación neural en el otro 4.
Se reporta el caso, debido a su rareza y similitud morfológica con otros tumores cutáneos, el PNET puede ser clínicamente y patológicamente subdiagnosticado; se debería incluir en el diagnóstico diferencial, incluso en casos simples, incluyendo estudio anatomo patológico.
Presentación del caso
Varón de 13 años, mestizo, natural de Huánuco- Perú, que acudió a consulta con dos tumores superficiales, ubicados en el vertex: el más grande de 3,5 cm. de diámetro del tono de piel del paciente, tenía una depresión en el centro por haber sido punzada con anterioridad y la segunda de 2 cm. de diámetro, de color eritemato-violáceo, de consistencia gelatinosa, ligeramente móviles y dolorosos a la palpación, con rápido aumento de volumen en cuatro meses (figura 1).
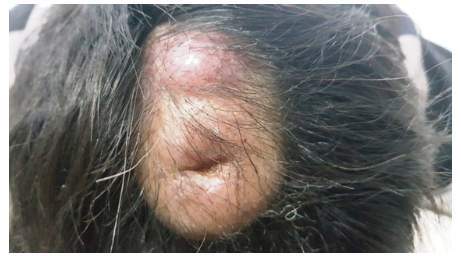
Figura 1 Tumores superficiales en cuero cabelludo; de 3,5 cm y 2 cm, con rápido aumento de volumen en 4 meses.
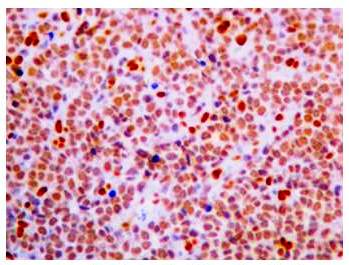
Se realizó biopsia por losange del tumor 2 cm (más pequeño), cuya descripción histológica fue, epidermis sin mayores alteraciones y en dermis presencia de una proliferación de células pequeñas redondas y azules de disposición nodular que ocupa predominantemente la dermis reticular y abarca hasta TCSC (figura 2 (A y B), figura 3). Se realizó examen inmunohistoquímico FLI1 (friendleukemia virus integration 1), donde se aprecia sobre expresión nuclear de FLI-1 (figura 4).
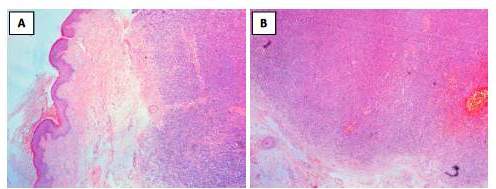
Figura 2 A: 40X, tinción hematoxilina-eosina. Epidermis sin mayores alteraciones y en la dermis presencia de una proliferación de células pequeñas redondas y azules de disposición nodular que ocupa predominantemente la dermis reticular y abarca hasta TCSC. B: 40X, tinción hematoxilina-eosina. En la dermis hay presencia de una proliferación de células pequeñas redondas y azules de disposición nodular que ocupa predominantemente la dermis reticular y abarca hasta TCSC.
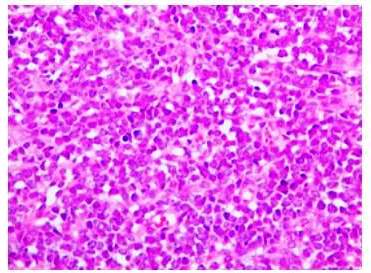
Figura 3 400X, tinción hematoxilina-eosina. Imagen a mayor aumento de la proliferación celular en donde se observa con mejor detalle el escaso citoplasma y la uniformidad entre ellas.
Discusión
La familia de tumores del sarcoma de Ewing (ESFT) representa una familia de neoplasias morfológicamente similares, incluyendo el sarcoma de Ewing clásico (ES) del hueso, el ES extra esquelético, el tumor de células pequeñas de la región toraco-pulmonar (tumor de Askin) y tumores neuro ectodérmicos primitivos del tejido blando (PNET) 5.
El sarcoma de Ewing extraóseo/tumores neuro ectodérmicos periféricos (ESS / PNET), son neoplasias raras que representan aproximadamente de 10 a 15% de los sarcomas de tejidos blandos en niños y el 5% de los sarcomas de tejidos blandos en adultos 6. Clínicamente, morfológica y genéticamente ESS/ PNET, comparten muchas características, lo que respalda la hipótesis de que estas dos neoplasias están histogenéticamente relacionadas y son consideradas como parte de la misma familia de tumores 7.
Los sitios extra esqueléticos comúnmente afectados incluyen los espacios paravertebrales, extremidades inferiores, cabeza, cuello y pelvis; otras ubicaciones raras de EES incluyen el retroperitoneo, epiplón, órbita, piel y pared torácica; es más probable que surjan tumores extra esqueléticos desde ubicaciones axiales y es menos probable que aparezcan de la pelvis 8.
El cuadro clínico es poco común., usualmente involucra tejidos subcutáneos o músculos profundos, y rara vez ocurre como cáncer de piel primario; se presenta como una tumoración superficial de 2-3 cm, de consistencia blanda, móvil, a veces dolorosa; el tumor se encuentra en la dermis media o profunda o tejido superficial subcutáneo y puede involucrar a la papila dérmica, con presentación tumoral pedunculada (9).
El diagnóstico diferencial se realiza con otras neoplasias compuestas de células redondas azules pequeñas: neoplasias primarias y metástasis cutáneas; las neoplasias primarias pueden ser: carcinoma de células de Merkel, espiradenoma ecrino, linfomas, sarcoma de células claras, rabdomiosarcoma, tumor rabdoideo maligno, tumor neuro ectodérmico primitivo maligno, carcinoma mioepitelial, histiocitoma fibroso angiomatoide, tumores anexiales pobremente diferenciados y sarcoma granulocítico; y las metástasis cutáneas pueden derivar de: sarcoma de Ewing óseo, carcinoma neuroendocrino de células grandes, carcinoma de pulmón de células pequeñas y neuroblastoma 9.
El diagnóstico puede requerir varias técnicas auxiliares tales como citología por aspiración, tinciones histoquímicas, inmunohistoquímica, microscopía electrónica, citogenética y genética molecular de traslocaciones; el aspecto histológico de células redondas pequeñas, positivo para CD99 en característico patrón membrana en la histoquímica y la translocación cromosómica específica que implica genes EWSR1 en el cromosoma 22q12 son criterios esenciales para diagnóstico de EES 10.
El tratamiento se realiza inicialmente con resección quirúrgica, asociado o no a la quimioterapia o radioterapia; según tamaño y ubicación del tumor, la radioterapia se usa cuando hay márgenes positivos después de la resección quirúrgica 11. Por lo tanto, para la enfermedad se requiere una estrategia terapéutica multimodal, agresiva 12. En pacientes con enfermedad localizada se estima que la tasa de supervivencia a 5 años es alrededor de 70%, debido al considerable progreso de la terapia tanto local como sistémica, durante las últimas 4 décadas 13.
El carácter menos agresivo del cuadro clínico cutáneo probablemente se produce debido a la ubicación superficial, tumores más pequeños y de fácil acceso, permitiendo la detección a través del examen clínico o por el propio paciente, diagnóstico precoz y resección quirúrgica completa, evitando el inicio de metástasis 10. Cerca de 30-40% de pacientes con ESS/PNET sufren de tumores recurrentes y tienen un muy mal pronóstico, la supervivencia a 5 años después de la recurrencia de estos pacientes fue de a 10-15%, y de 7% para aquellos cuya enfermedad reapareció en 2 años 7.
Una gran cohorte de pacientes con sarcoma de Ewing localizado tratado con modernos protocolos de quimioterapia, determinaron que la edad avanzada, la raza no blanca y la VSG basal elevada son factores pronósticos adversos independientes en pacientes con EES, y que el patrón de recaída no difiere entre EES y ES esqueléticos 14.
Antes del uso de la terapia sistémica multiagente, la tasa de supervivencia a largo plazo, con cirugía o radiación era inferior al 10%; con la adición de la quimioterapia multi- farmacológica para la cirugía o radiación, las tasas de supervivencia a 5 años varían de 49 a 60% en pacientes con enfermedad localizada, siendo los regímenes de quimioterapia estándar actual: vincristina, doxorrubicina y ciclofosfamida, alternando con ifosfamida y etopósido (régimen VAC-I / E) para la enfermedad localizada, y vincristina, doxorrubicina y ciclofosfamida (régimen VAC) en la enfermedad metastásica 15.
Aunque son tumores raros, los EES/PNET, se deben considerar en el diagnóstico diferencial, una adecuada evaluación clínica, estudios histológicos, inmunohistoquímicos y citogenéticos, realizados a tiempo y un tratamiento apropiado resultan fundamentales para su correcto manejo, debido a que la enfermedad cutánea, generalmente, presenta un curso lento y un pronóstico favorable.

