











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La hemostasia es un mecanismo de defensa cuya finalidad es conservar la integridad y permeabilidad vascular para evitar la pérdida de sangre tras una lesión, la cual se puede comprometer por anormalidades hereditarias o adquiridas 1-4, siendo una de ellas, la enfermedad von Willebrand, que corresponde el defecto hereditario más frecuente a nivel de la hemostasia primaria (1, 4-9).
Los pacientes con alteraciones en la hemostasia constituyen un grupo de riesgo en Odontología, por lo que es importante entenderlas, así como conocer su impacto en el manejo del paciente, ya que muchas de estas pueden iniciarse con un sangrado persistente en cavidad oral de origen desconocido 1,10. Por tanto, al ser la enfermedad von Willebrand, el desorden hemorrágico más frecuente 1,11, es pertinente reforzar al odontólogo el conocimiento práctico de este trastorno, mediante la revisión de artículos de referencia relevantes, dando énfasis, de una forma concisa y precisa, las características y pruebas diagnósticas de la enfermedad von Willebrand, tratamiento de esta enfermedad, pautas previo al tratamiento odontológico y recomendaciones durante el manejo del paciente en odontología.
Factor von Willebrand
El factor von Willebrand es una glicoproteína multimérica que se encuentra circulando en plasma, necesaria para la adhesión plaquetaria en el subendotelio vascular dañado, lo que favorece, además, la formación del coágulo (1,3,4,7,9,10,11,12,13,14,15). Asimismo, forma un complejo con el factor VIII (1,3,4,7,8,9,10,11,12,13,14,15,16).
Enfermedad von Willebrand
La enfermedad von Willebrand es el desorden hemorrágico hereditario más común, que se origina por la deficiencia del factor von Willebrand, la cual provoca una adhesión y agregación plaquetaria defectuosa (1,2,3,5,6,10,11,12,13,14,15). El defecto puede ser cuantitativo, es decir, que existe deficiencia del factor, o cualitativo, lo cual se refiere a disfunción del factor von Willebrand (2,3,4,5,7,8,9,10,11,12,13,14,15,17).
Su prevalencia, en la población general, se estima en un rango de 0.6% a 2%, pero se debe de considerar que existe aproximadamente un 3% de personas que pueden ser portador asintomático (1,2,6,7,10,14). Su transmisión es autosómica dominante, aunque también se ha descrito herencia recesiva. No tiene predilección sobre género (3,4,5,6, 9,10,11,12,13,17).
Se caracteriza por un tiempo de sangrado y tiempo parcial de tromboplastina (TPT) prolongado, con bajos valores del factor VIII, y aumento de fragilidad capilar, pero con un recuento de plaquetas normal (1,3,5,6,7,13,14).
Clasificación
La enfermedad von Willebrand se clasifica en 3 categorías principales. En la Tabla 1 se detallan los tipos y sus principales diferencias (2,4,5,7,9, 11,12,13,14,15,16, 17).
Tabla 1 Clasificación de la enfermedad von Willebrand y sus características.
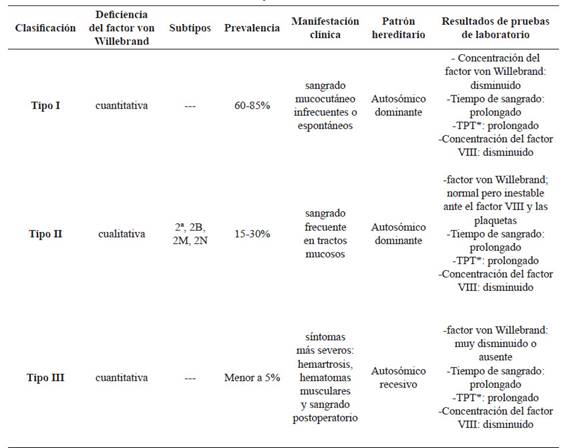
*TPT: tiempo de protrombina
Diagnóstico
El diagnóstico de la enfermedad von Willebrand, puede ser un reto ante el número de pruebas de laboratorio para el diagnóstico, ante la interpretación debido a la heterogeneidad de la enfermedad, y ante la variación fisiológica en los niveles del factor von Willebrand y factor VIII (2, 4).
La elaboración de una correcta y detallada historia clínica, incluyendo la historia personal y familiar de sangrado excesivo es importante para el diagnóstico y manejo del paciente, así como la interconsulta previa con el hematólogo para delimitar las características de la enfermedad y pautas de tratamiento, y precisar, por último, la planificación adecuada del tratamiento a realizar (1,2,3,4,5,6,9,10,13,15,18,19). Esta historia clínica debe ir acompañada, además, de pruebas de laboratorio para definir el fenotipo de la enfermedad, pues determinarán si el defecto del factor von Willebrand es cualitativo o cuantitativo, ya que la severidad de la tendencia de sangrado es usualmente proporcional al grado de deficiencia primaria del factor von Willebrand y a aquella deficiencia secundaria del factor VIII (4,5,9, 4), siendo estas las siguientes:
Factor von Willebrand y co-factor de ristocetina (FvW:RiCo) conocida también como actividad de factor von Willebrand: Esta prueba es la piedra angular en el diagnóstico de la enfermedad, pues evalúa la actividad del factor von Willebrand, ya que se basa en la propiedad de factor von Willebrand de aglutinar plaquetas en presencia de ristocetina in vitro (conocido como RIPA, abreviatura en inglés de: Ristocetin-Induced Platelet Aggregation) y cuantificar la aglutinación producida (2,4,6, 7,10,11,13,14).
Antígeno del factor von Willebrand 13.
Análisis de los multímeros del factor von Willebrand en plasma 4,7,11,13.
Factor VIII intraplaquetario 6,10,14.
Determinación de la actividad de factor VIII: mide la actividad coagulante en el contexto de la evaluación de la enfermedad von Willebrand, es reflejo indirecto de la capacidad del factor von Willebrand de unirse al factor VIII y estabilizar su vida media 4.
Unión factor von Willebrand - colágeno: valora la capacidad de factor von Willebrand plasmático de unirse al colágeno 4.
Unión factor von Willebrand - factor VIII: evalúa la habilidad del factor von Willebrand a unirse a factor VIII normal (exógeno) (4, 7).
Factor von Willebrand plaquetario 7.
Anticuerpos anti-factor von Willebrand 4.
Tiempo de sangrado: determina la función de plaquetas (rango normal 1-7 minutos), es decir, permite conocer la calidad de plaquetas. Su valor normalmente es prolongado 1,3,4,6,7,11.
Tiempo de trombina, el cual es normal 4,6,7,18.
Tiempo parcial de tromboplastina: determina el patrón de coagulación intrínseca (rango normal: 25 ±10 segundos) 1,3,4,11,13.
Proporción internacional normalizado para medir el factor extrínseco: el rango normal es 1.0. Se define como la proporción entre el tiempo de protrombina, en segundos, del paciente y un control de tiempo de protrombina estandarizado, por medio del así llamado Índice de Sensibilidad Internacional 1,11.
Analizador de la función plaquetaria - PFA (abreviatura en inglés de Platelet Function Assay): se usa para medir la capacidad que tienen las plaquetas para cerrar una herida en una membrana (4, 7).
Conteo de plaquetas: cuantifica la función plaquetaria (rango normal 150.000 - 450.000/ul), el cual usualmente es normal 3,4,6,7,10,11,14).
Conteo total de sangre: determina la anemia por deficiencia de hierro, la cual se puede desarrollar como consecuencia de la pérdida de sangre crónica y de la trombocitopenia 13.
Tiempo de sangrado capilar: permite valorar la severidad del desorden de sangrado, aunque no es una prueba útil para la enfermedad tipo I von Willebrand por tener valores dentro de los límites normales 13.
Grupo sanguíneo 13.
Ensayos de fibrinógeno, actividad del factor IX, prueba de inhibidores 6.
Exámenes de hepatitis B y C, chagas, sífilis y VIH, ya que se puede requerir de suministro plasmático en estos pacientes 6).
Propéptido factor von Willebrand 4).
Respuesta al DDAVP (1-desamino-8-D-arginina vasopresina) del paciente 4
A pesar de que se mencionan numerosas pruebas de laboratorio, el FvW-Antigénico (FvW-Ag) y el co-factor de ristocetina (FvW:RiCo) son las pruebas elementales para el diagnóstico de la enfermedad von Willebrand. Adicionalmente, existen pruebas de laboratorio cuya función radica en la adecuada subclasificación de la enfermedad von Willebrand, con el fin de poder brindar un tratamiento preciso (estudio de multímeros y la agregación plaquetaria inducida por ristocetina, es decir RIPA) 7).
Manifestaciones clínicas
La enfermedad von Willebrand se caracteriza por una tendencia de hemorragia de leve a moderada con un patrón de sangrado mucocutáneo, además, puede presentar hematomas, epistaxis, menorragia, hematuria, sangrado, sea gingival, postoperatorio excesivo o después de extracciones dentales; en casos severos puede tener un patrón de sangrado hemofílico con aparición espontánea en articulaciones y músculos (hemartrosis o hemorragias musculares) (1, 2, 5,6,7,8,9,10,11,12,13,14).
Tratamiento
El objetivo de tratamiento para la enfermedad von Willebrand es corregir el defecto dual de hemostasia, es decir, la adhesión plaquetaria anormal y la coagulación anormal, debido al bajo nivel de factor VIII, con el fin de aumentar los niveles de plasma del factor von Willebrand y factor VIII (5,7,9,12,14,15). Existen dos principales opciones terapéuticas disponibles: la desmopresina, que libera el factor VIII endógeno de las células endoteliales, y la terapia de reemplazo con concentrados de factor von Willebrand conteniendo factor VIII 7,12,14,15). Para determinar cuál es la estrategia más útil, dependerá de la respuesta individual a la terapia de desmopresina así como a la ausencia de sangrado y los niveles basales del factor von Willebrand y factor VIII funcionales 2,4,7,12,13,14,15). Otras formas adicionales de tratamiento, a los previamente mencionados son los concentrados de plaquetas, inhibidores sintéticos de fibrinólisis, preparaciones orales de anticonceptivos, entre otros (7, 12).
Desmopresina
La desmopresina (1-desamino-8-D-arginina vasopresina), es un análogo sintético de la vasopresina, de la hormona antidiurética vasopresina, que fue originalmente diseñada para el tratamiento de la diabetes insípida, la cual estimula la liberación del factor von Willebrand de las células endoteliales con el fin de aumentar las concentraciones circulantes del factor von Willebrand y factor VIII, así como aumentar la adhesión plaquetaria 1,3,5,7,9,10,12,13,14.
El principal tratamiento para pacientes con la enfermedad von Willebrand es el reemplazo del factor de coagulación deficiente en la circulación, es decir, la desmopresina, pues ayuda a prevenir eventos hemorrágicos en aquellas personas con enfermedad von Willebrand tipo 1 y algunos tipo 2, pues la segunda alternativa sería la infusión de concentrado de derivados de plasma, conteniendo el factor VIII y el factor von Willebrand, por tanto 20% aproximadamente de los pacientes no responden a la desmopresina (1, 3, 5, 9, 12,13,14,15,16, 17).
Entre sus ventajas se señalan la seguridad, ya que evita riesgos de transmisión sanguínea de virus o nuevos patógenos así como la precoz aloinmunización plaquetaria inherente a la terapia de reemplazo, además, otras ventajas son su efectividad y el bajo costo 7,9,12,13,14,16.
Está contraindicada en pacientes con tipo 2B de la enfermedad von Willebrand, ya que el aumento de las concentraciones del factor von Willebrand pueden inducir a trombocitopenia severa a consecuencia de la unión plaquetaria excesiva así como a trombosis 3,9,12,14,15,16,17). Los pacientes con tipo 3 de la enfermedad no responden a la terapia con desmopresina, por lo que se debe de buscar otra alternativa 3,5,9,12,13,14,15,16,17).
Otra contraindicación, es la taquifilaxis, que se presenta posterior a infusiones diarias repetidas de desmopresina, provocando ausencia de respuesta al tratamiento. En estos casos o para aquellos pacientes donde la desmopresina está contraindicada, el factor von Willebrand debe ser reemplazado mediante productos sanguíneos 5,9,13,14).
Los efectos colaterales de la desmopresina son característicos, aunque leves, transitorios y raros, estos incluyen rubor, taquicardia, disminución de la presión sanguínea, náusea y dolor de cabeza, así como calambres abdominales 5,9,12,13,14,15). Estos síntomas son atribuidos a los efectos vasodilatadores de la droga y pueden ser atenuados por la administración paulatina de la infusión 12). Existen, también, complicaciones más serias como convulsiones, hiponatremia debido al efecto antidiurético de la droga y trombosis, no obstante, esta se presenta solo en pacientes con otros factores de riesgo trombótico, por ejemplo, pacientes mayores de 70 años o pacientes con historia de enfermedad cardiovascular, arterioesclerosis, hipertensión u otros 5,8,9,12,13,14,15).
Terapias transfusionales
Los productos sanguíneos que se administran en pacientes con desórdenes hemorrágicos congénitos o adquiridos tienen la desventaja de asociarse con contaminantes virales como el virus del sida, el virus de la hepatitis; e igualmente, puede haber efectos colaterales como anemia hemolítica, formación de factores inhibitorios y reacciones alérgicas, sin embargo, en la actualidad los productos recombinantes (no derivados del plasma) reducen el riesgo 3,7,8,9).
El factor VIII y factor von Willebrand pueden ser transfundidos como plasma fresco congelado, pero el gran volumen que se requiere limita su uso, además de que existe la posibilidad de la presencia de todos los efectos adversos ya previamente mencionados 1,7,8,12).
El crioprecipitado es una terapia transfusional. Es un concentrado intermedio viralmente inactivado de factor VIII, que contiene de 5 a 10 veces más factor VIII y factor von Willebrand que el plasma fresco congelado 3,9,12,13,15,16). Se utiliza posterior a la administración de desmopresina cuando las cantidades de factor von Willebrand y factor VIII son insuficientes, o si todos los demás tratamientos han fracasado; inclusive a veces, es necesario la transfusión de concentrados de plaquetas (5, 9,10,12,13,15,16). Específicamente, se emplea en pacientes con enfermedad von Willebrand severa (severa tipo 1, tipo 3 y tipo 2B), en pacientes con sangrado severo, con historia de respuesta desconocida a la desmopresina, cuando la desmopresina esté contraindicada o existe amenaza de vida (1).
Antifibrinolíticos
El tratamiento con desmopresina es importante para el control del sangrado, pero no es exclusivo, ya que los métodos locales y mecánicos son necesarios también, es decir, que los agentes antifibrinolíticos, aunque no pueden mejorar de forma aislada el sangrado, son coadyuvantes, efectivos y baratos, como alternativas para el control de la hemorragia, y protección del coágulo, además de que la saliva refuerza la función debido a su acción fibrinolítica 2,3,8,10,12,13,15,16,17,19).La función de un agente antifibrinolítico se basa en retardar la disolución de fibrina mediante la inhibición de sustancias activadoras de plasminógeno 2,5,8). Ejemplos de estos agentes son el ácido tranexámico, e-ácido aminocapróico, y el coágulo de fibrina 2,8,10,15,16). Todos los agentes pueden ser usados antes, durante o después de los procedimientos odontológicos, vía oral o vía intravenosa (1,2,10,12,15,16), pero están contraindicados en casos de que el paciente curse con una enfermedad trombótica activa 8).
Entre las características de los agentes antifibrinolíticos mencionados se encuentran:
Selladores, pegamento o adhesivos de fibrina: es un material que contribuye al sanado de la herida, por ejemplo, en casos de extracciones dentales o cirugías maxilofaciales, que contiene derivados de plasma y que ayuda a la coagulación, convirtiendo el fibrinógeno en fibrina, pues tiene propiedades hemostáticas que actúan independientemente del patrón de coagulación del paciente, aunque su función se puede disminuir por el efecto sistémico de la heparina 10,13,16,19).
Ácido tranexámico: es un compuesto antifibrinolítico que se deposita en los sitios de unión sobre el plasminógeno y la plasmina, bloqueando la unión de la plasmina a la fibrina, por lo que inhibe la fibrinólisis, y estabiliza el coágulo. Su uso es exitoso en cualquier zona de sangrado 5,10).
Trombina tópica: es un material que se utiliza directamente en el sitio sangrante; en cavidad oral se dificulta su uso debido a la presencia de sangre, saliva y los movimientos musculares 10,19).
Consideraciones odontológicas
El tratamiento odontológico en un paciente con enfermedad von Willebrand, debe ser individualizado de acuerdo a la severidad de la condición de este, y coordinado con el hematólogo, quien debe de determinar el tipo de enfermedad y la necesidad del reemplazo del factor de coagulación según diagnóstico específico, con el fin de definir el régimen profiláctico apropiado para evitar el sangrado local durante las intervenciones orales, según el procedimiento dental a realizar 1,2,3,6,15,18,19).
Es conocido que los pacientes con desórdenes de coagulación pueden presentar problemas odontológicos severos, como por ejemplo, caries severa y problemas periodontales, debido al temor que ellos pueden tener por la presencia de sangrado durante el cepillado, siendo por tanto, que el problema oral no sea origen de la enfermedad sistémica sino del grado de higiene oral, así como por el descuido ante una atención preventiva y primaria 3,10).
Ante un paciente con enfermedad von Willebrand, el profesional odontólogo debe de valorar, como se mencionó anteriormente, el tipo de desorden hematológico hereditario involucrado y el tratamiento hematológico a recibir, asimismo, debe de evaluar posibles modificaciones de dicha terapia y el alcance que pueda tener para el tratamiento odontológico a ejecutar; análisis de las drogas que puedan ser contraindicadas, vida media del factor transfundido, lo que es esencial para definir el tipo de tratamiento a realizar y el tiempo de duración disponible para trabajar, así como la posibilidad de riesgo de sangrado ante cualquier tratamiento dental invasivo 17).
En la literatura se destaca que la incidencia de sangrado postoperatorio después de tratamientos odontológicos invasivos, llevando a cabo las medidas de hemostasia correspondiente se estima entre 0,2% y 3,3%, mientras que en pacientes con desórdenes de hemostasia tiene un rango de 8,6%-32,1% 17).
Entre las consideraciones especiales para efectuar el tratamiento odontológico en un paciente con enfermedad von Willebrand se encuentran:
Definición y organización del plan de tratamiento por sesión con el fin de minimizar el trauma en cavidad oral (2, 9)
Elección cuidadosa de técnicas de anestesia, para evitar la formación de hematomas y, por consiguiente, obstrucción de la vía aérea, lo cual puede suceder con la técnica para el bloqueo dentario-inferior, o anestesia troncular 2,3,13,16. Se recomienda el uso de la anestesia local, ya que el único lugar de hemorragia sería en el lugar de la extracción, facilitando el control del sangrado con hemostáticos locales o en dado caso, con métodos sistémicos de control de hemorragia 2,16.
precauciones ante la intubación traqueal o el uso de una máscara laríngea durante un procedimiento bajo anestesia general, para así evitar un hematoma laríngeo 2,16.
Utilización de técnicas modificadas, minuciosas, detalladas y/o refinadas quirúrgicas para reducir la hemorragia durante un procedimiento dado 1,2,5,18).
Selección y delimitación del número de dientes a extraer en una cita 2).
Permitir la exfoliación de los dientes temporales según el tiempo que les corresponda 2,3
Inducción a la formación y estabilización del coágulo en casos de extracción dental, para evitar el sangrado postoperatorio, ya que un coágulo fuerte y estable es la mejor hemostasia 16,19,20).
Remoción, en el caso dado, del coágulo exofítico que se forme en el alvéolo, para evitar sangrado contínuo y obstrucción de compresión en la zona 2,6).
Utilización de medidas locales junto con las terapias establecidas por el hematólogo, para evitar el sangrado en procedimientos dentales invasivos, sea perioperatorio o postoperatorio 17). Por lo anterior, se hace pertinente el conocimiento de diferente conductas de manejo de sangrado así como materiales, por ejemplo, uso de suturas (preferiblemente reabsorbibles), compresión local con gaza, uso de férula para proteger el coágulo, placas preformadas, agentes químicos (trombina) o productos hemostáticos reabsorbibles (celulosa oxidasa regenerada, colágeno microfibrilar, esponjas de fibrina o coágulos de gelatina), uso de antifibrinolíticos así como uso de electrocauterio, entre otros 1,2,3,5,6,8,10,12,13,16,17,19,20).
Utilización de analgésicos como Acetaminofén o medicamentos opiáceos que no alteren la función plaquetaria, como sucede con los medicamentos Aines y el ácido acetil salicílico 6,13,20).
Uso de materiales que permitan el aislamiento del campo de trabajo en operatoria dental, para evitar el daño de los tejidos blandos con los instrumentos cortantes, por ejemplo, el dique de hule (3, 6), así como mantener el cuidado pertinente con la utilización de diferentes materiales odontológicos de rutina, sean radiografías, bandas, entre otros 10).
Realización de tratamiento de endodoncia, en un diente a extraer cuando sea posible, para evitar el sangrado del alvéolo 2,10).
Realización de raspado y curetaje periodontal, bajo el uso postoperatorio de antifibrinolíticos 3).
Consideración de la posibilidad de administrar, ante sangrado localmente en la zona intervenida, anestésico local con epinefrina, para favorecer la hemorragia lenta, la vasoconstricción, la formación del coágulo y su estabilización, sin descartar que después del efecto vasoconstrictor (epinefrina) localmente, puede acontecer vasodilatación y consecuentemente hemorragia 2).
Valoración de la ejecución del procedimiento quirúrgico en hospital, cuando el caso lo amerite 1).
Promoción de constante educación, prevención y mantenimiento de buena higiene oral 1,6,10,12,13).
Síndrome adquirido de von Willebrand
Es un trastorno hemorrágico adquirido que asemeja la enfermedad von Willebrand, en cuanto a los resultados del laboratorio, pues presenta un tiempo de sangrado prolongado y bajos niveles en plasma del factor VIII y von Willebrand, y hallazgos clínicos similares, como sangrado mucocutáneo y/o gastrointestinal. Su prevalencia se estima en 0,04% 4). Es importante mencionar que la deficiencia del factor von Willebrand es secundaria a síndromes linfoproliferativos o mieloproliferativos, enfermedades cardiovasculares o enfermedades malignas, desórdenes autoinmunes, o menos frecuente hipotiroidismo, uremia, e incluso en algunos casos su origen es desconocido 4).
DISCUSIÓN
Los profesionales en odontología deben tener el conocimiento en cuanto al manejo y tratamiento de los desórdenes hemorrágicos, en este caso la enfermedad von Willebrand, previo a realizar cualquier tipo de atención odontológica. La mejor forma de evitar complicaciones hemorrágicas, tras procedimientos orales, es la prevención y para ello es indispensable disponer de una historia clínica detallada del paciente, lo que significa tener un enfoque de tratamiento temprano y efectivo, pues el diagnóstico y manejo de estos pacientes dependerá del conocimiento de los mecanismos normales de la hemostasia y su análisis, así como de los procedimientos simples para el control de la hemorragia, sin obviar la participación del paciente ante el cuidado dental y medidas profilácticas pertinentes, quien tiene que contribuir activamente para mantener una buena condición oral.
El profesional en odontología debe de trabajar en equipo, específicamente con el hematólogo, lo que obliga una comunicación responsable para valorar el riesgo y definir de forma conjunta el plan de tratamiento apropiado para el paciente, con el fin de minimizar las condiciones propias del trastorno, ante un diagnóstico exacto individualizado del tipo de enfermedad von Willebrand y la necesidad del tratamiento, específico y coordinado.
El riesgo de sangrado en esta población existe, y se debe de ser consciente de ello, lo que conduce a la completa preparación del profesional, para poder brindar la mejor solución de tratamiento, en un tiempo dado considerando todos los factores.

