










Servicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Gastroenterología del Perú
versión impresa ISSN 1022-5129
Rev. gastroenterol. Perú v.22 n.3 Lima jul./set. 2002
TRABAJOS ORIGINALES
Gastritis crónica atrófica: mecanismos patogénicos por hipersensibilidad celular
Rosemary Recavarren Asencios1; Sixto Recavarren Arce1
1 Servicio de Patología de la Clínica Ricardo Palma y
Departamento de Patología del Hospital Nacional Cayetano Heredia
RESUMEN
La Gastritis Crónica Atrófica, de alta incidencia en la infección por Helicobacter pylori, no ha tenido hasta hoy mecanismos patogénicos establecidos.
La Gastritis por Helicobacter pylori, claramente relacionada a la Gastritis Crónica Activa Superficial, no ha encontrado mecanismos que la conecten con la Gastritis Crónica Atrófica. Hasta hace poco, eran consideradas por algunos autores como lesiones independientes.
En el presente trabajo, se estudiaron 42 biopsias gástricas antrales o corporales, con Gastritis Crónica Atrófica en diferentes estadíos. Las lesiones histológicas que comprenden a esta última, van desde la "infiltración linfoide profunda" en las glándulas gástricas propias, hasta el reemplazo de las mismas por tejido fibroinflamatorio.
Por métodos inmunohistoquímicos, hemos podido comprobar que las células linfoides que infiltran el estrato glandular propio del estómago están conformadas por linfocitos T CD8+ (citotóxicos) y por células linfocíticas B secretoras de anticuerpos. Planteamos en el presente trabajo que, los linfocitos T citotóxicos agredirían a las glándulas propias antrales y corporales destruyéndolas y produciendo de esta forma su reemplazo por tejido fibroinflamatorio. Similares acciones se producirían por linfocitos B, pero a través de la secreción de anticuerpos locales contra células epiteliales glandulares gástricas.
Palabras Claves: Helicobacter pylori, imunohistoquímica, gastritis atrófica, linfocitos T citotóxicos.
SUMMARY
Chronic Atrophic Gastritis of high incidence in Helicobacter pylori infection has not been pathogenically explained yet.
Helicobacter Pylori Chronic Active Superficial Gastritis has not been related to Chronic Atrophic Gastritis by some authors, who considered, until some time ago, that they both were independent lesions. In this study, we examined 42 antral or corporal gastric biopsies with Chronic Atrophic Gastritis at different stages, with histological lesions going from "deep lymphoid infiltration" in the proper gastric glands, to the replacement by fibro inflammatory tissue.
With immunohystochemistry methods we have been able to prove that the lymphoid cells infiltrating the glandular part of the gastric mucous membrane are composed by CD8+(cytotoxic)T lymphocytes and by B lymphocites antibody secretors. In this study we suggest that cytotoxic T lymphocytes damage and destroy antral and corporal gastric proper glands, with further fibro inflammatory tissue replacement. Similar actions would be produced by B lymphocytes, but by secreting local antibodies against the cells from proper gastric glands.
Key Words: Helicobacter pylori, Inmunohystochemistry, atrophic gastritis, cytotoxic T lymphocyte.
INTRODUCCIÓN
Los mecanismos patogénicos que producen la gastritis crónica atrófica (GCA), condición gástrica considerada como premaligna1,2, no han quedado hasta hoy bien establecidos. Las lesiones inflamatorias agudas que dañan los cuellos glandulares de la mucosa gástrica conocidos como "neck lesion" (lesiones del cuello), han sido consideradas como las alteraciones histológicas más importantes en la génesis de la atrofia de las glándulas propias del estómago1,2. Sin embargo, esta propuesta no explica con suficiente claridad las diferentes características histológicas que corresponden a la GCA.
En el presente estudio, se propone que en las lesiones producidas por Helicobacter pylori (Hp) en la mucosa gástrica y que conllevan a la GCA, existen dos etapas. En la primera etapa, se ha observado que la lesión primordial se produce en el epitelio de cubierta protector del estómago, esta lesión que es producida por acción enzimática bacteriana estimula una reacción inflamatoria de tipo infeccioso que característicamente afecta sólo la porción superficial de la mucosa gástrica comprometiéndola hasta los cuellos glandulares. Esta alteración es diagnosticada por los patólogos como gastritis crónica activa superficial por Helicobacter pylori; el componente celular está predominantemente conformado por neutrófilos, células plasmáticas y linfocitos2,5.
La segunda etapa de la gastritis por Helicobacter pylori, se caracteriza por producir además inflamación crónica de las glándulas antrales y/o corporales. Esta lesión es diagnosticada por los patólogos con diferentes nombres tales como: Gastritis crónica profunda, intersticial o difusa. Esta inflamación crónica es a predominio de linfocitos, algunas células plasmáticas y eosinófilos y característicamente ausencia de neutrófilos.2-5
En la gastritis crónica profunda, hemos observado que los linfocitos en múltiples áreas se encuentran en íntimo contacto con las células epiteliales de la mucosa gástrica. Esta lesión ha sido denominada en el presente estudio como "Lesión linfocítica epitelial".
Las lesiones encontradas en el presente estudio en la gastritis crónica atrófica son muy similares a las observadas en Tiroiditis de Hashimoto19, Enfermedad de Sjögren 20 y Hepatitis crónica del tipo viral, enfermedades por hipersensibilidad celular. De acuerdo a este concepto se propone que la Gastritis Crónica Atrófica, sea también producida por agresión a epitelios parenquimatosos por células linfoides hipersensibles.
Para comprobar esta propuesta, se han realizado estudios de inmunohistoquímica en las biopsias de pacientes con Gastritis Crónica Atrófica, que han permitido detectar mecanismos inmunes de hipersensibilidad que producen alteraciones de las glándulas propias gástricas y que a su vez conducen a la GCA.
Otra alteración histológica observada en el presente estudio, consiste en la presencia de agregados linfoides que involucran o reemplazan a glándulas gástricas propias, los cuales han sido diferenciados de folículos linfoides.
MATERIAL Y MÉTODOS
Para la realización del presente estudio, se seleccionaron 42 biopsias gástricas con gastritis crónica profunda.
Las biopsias provienen de series utilizadas en proyectos de investigación en Gastritis por Helicobacter pylori del Hospital y Universidad Peruana Cayetano Heredia y de la Clínica Ricardo Palma.
(I) ESTUDIOS HISTOLOGICOS CON HEMATOXILINA Y EOSINA
Se estudiaron secciones histológicas coloreadas con Hematoxilina y Eosina en los 42 casos, las secciones tuvieron 4 a 6 micras de espesor. En el estudio histopatológico se determinó los siguientes tipos de alteración estructural:
1. Gastritis Superficial: caracterizada por proceso inflamatorio crónico activo superficial (compromiso inflamatorio hasta los cuellos glandulares).
2. Gastritis Profunda: caracterizada por proceso inflamatorio a células redondas y eosinófilos en el estrato de glándulas propias del antro o cuerpo gástrico.
3. Detección de lesión linfocítica epitelial: caracterizada por presencia de linfocitos adherentes a las células epiteliales de las glándulas gástricas; las que mostraban diferentes grados de alteración estructural y citológica. La graduación de la lesión se realizó cualitativamente tomando como grado leve, aquella en la que se observaba escasos linfocitos asociados a mínima distorsión estructural de las glándulas propias gástricas; siendo severa la lesión, ante la presencia de numerosos linfocitos asociados a lesión estructural de mayor magnitud. La lesión de grado moderado, se consideró en el intermedio entre los dos anteriores.
4. Agregados linfoides: caracterizados por cúmulos de células linfocíticas que presentaban o no eosinófilos en su estructura y también en algunos casos remanentes glandulares incorporados.
5. Áreas de atrofia fibroinflamatoria o fibrosa: en el estrato de glándulas propias gástricas.
6. Presencia de folículos linfoides diagnosticados por la presencia de cúmulos de linfocitos con o sin centro germinal y ausencia de eosinófilos o remanentes de glándulas propias gástricas.
7. Presencia de Metaplasia Intestinal superficial o profunda: en sus variados tipos histológicos.
8. Presencia de Helicobacter pylori: detectado en las coloraciones de Hematoxilina y Eosina y en los casos dudosos, por coloraciones de plata por Warthin-Starry.
ESTUDIOS CON INMUNOHISTOQUÍMICA
Los estudios de inmunohistoquímica se realizaron en el servicio de Patología de la Clínica Ricardo Palma. En las 42 biopsias se realizaron estudios de doble tinción por Inmunohistoquímica para la detección de linfocitos CD8+ citotóxicos y de estructuras epiteliales glandulares. Para ello se realizaron cortes histológicos de 4 a 6 micras de espesor que se extendieron en láminas polarizadas las cuales fueron manualmente teñidas con el Sistema EnVision TM Doublestain System ( DAKO Corp., Carpintería, CA) con anticuerpo monoclonal CD8+ (Anti-Human CD8+, T cell, Supresor/Cytotoxic, DAKO Corp., Carpintería, CA) y con el anticuerpo policlonal contra citoqueratinas ( Cytokeratin (Wide Spectrum Screening), DAKO Corp., Carpintería, CA); tomando como control de la tinción casos de biopsias con Tiroiditis de Hashimoto, las cuales son conocidas como poseedoras de células linfocíticas CD8+ en su estructura. Asimismo, en la detección de Linfocitos B, se realizaron tinciones manuales con el Sistema LSAB-2 (DAKO Corp., Carpintería, CA), para los 20 casos con anticuerpo monoclonal contra linfocitos B (Anti-Human CD20+, B cell, DAKO Corp., Carpintería, CA).
La lectura de los estudios por Inmunohistoquímica se catalogó como positiva,ante la evidencia clara de una tinción de membrana y/o citoplasma en las células con los marcadores CD8+ o CD20+ ; y de citoplasma, en el caso de la citoqueratina. La graduación de esta lectura se realizó cualitativamente en grado leve, cuando se evidenciaron escasos linfocitos CD8+ o CD20+ , asociados a células glandulares positivas a citoqueratina; en grado severo, cuando se evidenciaron abundantes linfocitos y en grado moderado, en el punto intermedio entre ambos.
RESULTADOS
Histología e Infección por Helicobacter pylori
De los 42 casos de pacientes incluidos en el presente estudio, 32 (74%) fueron positivos para Helicobacter pylori en tinción de Hematoxilina y Eosina, siendo 03 (7%), de estos casos, comprobados a través de la tinción Warthin-Starry ante la duda diagnóstica con la tinción de Hematoxilina y Eosina. El 100% presentó gastritis crónica profunda.
En la gastritis superficial, se determinó la presencia en grado moderado a severo de polimorfonucleares (56% de los casos), células plasmáticas (91% de los casos) y linfocitos (81% de los casos), por encima de los cuellos glandulares en la etapa inicial de la enfermedad.
La gastritis crónica profunda, está básicamente conformada por células mononucleares tipo linfocitos en grado moderado a severo en 41 (95%), de los casos, y de grado leve a moderado en 01 caso (5%), por debajo de los cuellos glandulares. En ningún caso se observó presencia de células polimorfonucleares en la infiltración profunda.
Los agregados linfoides, se observaron en 18 (42%), de los casos y los folículos linfoides, en 15 (32%), de los casos.
Lesión Linfocítica Epitelial
La lesión linfocítica epitelial, fue observada en 37 de los 42 casos (87%); de los cuales, en grado severo 8 (22%), moderado 17 (46%) y leve 12 (32%), de los casos.
Tinciones de Inmunohistoquímica
Los estudios de Inmunohistoquímica para detectar linfocitos citotóxicos CD8+ y Linfocitos B+, demostraron positividad en el 100% de los casos de gastritis crónica profunda y característicamente la positividad se dio sólo en los estratos profundos de la mucosa gástrica (ver fotografías 5 y 6). Solo en un caso (2%), se observó también leve reacción en la parte superficial de la mucosa gástrica. En el presente estudio, no se encontraron linfocitos T CD8+ intraepiteliales. La graduación de la positividad para linfocitos CD8+, fue de grado leve en 19 (45%), de los casos, moderado en 15 (36%) y severo en 8 (19%).
Las tinciones para linfocitos B, fueron de grado moderado a severo en todos los casos en la gastritis crónica profunda.
Los agregados linfoides tuvieron en su estructura linfocitos CD8+ y CD20+ , en el 100% de los casos. No se realizaron estudios de Inmunohistoquímica con otros marcadores.
DISCUSIÓN
La GCA, condición premaligna, no ha logrado hasta el momento explicación patogénica transparente1-6. La teoría aducida en la literatura acerca de la lesión de los cuellos glandulares ("neck lesion"), producida por acumulación de polimorfonucleares que conllevan a la destrucción del potencial regenerativo de la glándula2 es endeble desde el punto de vista histopatogénico; ya que un sólo túbulo glandular y sus ramificaciones irían a la atrofia, con un patrón histológico que no es el que se observa en la GCA. Otros mecanismos, probablemente los de hipersen sibilidad, estarían involucrados en la génesis de esta lesión3-5,10-18,22-24,26,29.
Aunque en la mucosa gástrica de un adulto normal existen escasos linfocitos en la lámina propia y también algunos intraepiteliales, esas células linfoides no ejercitan alteración de las estructuras normales del estómago.
La comprobación inmunohistoquímica de linfocitos agresores (T CD8+ citotóxicos y B), en los estratos profundos del proceso inflamatorio gástrico con demostración clara de destrucción del epitelio glandular y la consiguiente atrofia, son hallazgos suficientes para proponer un mecanismo inmune hipersensible como productor de la lesión glandular gástrica conducente a la gastritis atrófica. El hallazgo de las lesiones descritas en el presente estudio como agregados linfoides, constituiría una etapa avanzada de este proceso infiltrativo.
| Foto 1. - Daño Epitelial Mucoso producido por Helicobacter pylori. Se observa entre las flechas la destrucción de la porción mucinosa del epitelio de cubierta. Cabeza de flecha indica foveola con preservación de la mucina epitelial (X 400) Coloración Warthin-Starry. |
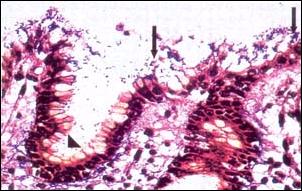
| Foto 2. - Gastritis Antral Superficial Crónica Activa de grado severo. El proceso inflamatorio solo se da en la parte superficial de la mucosa antral; las glándulas propias están preservadas. Al fondo muscularis mucosae (mm). (x 150) Coloración Hematoxilina y Eosina. |
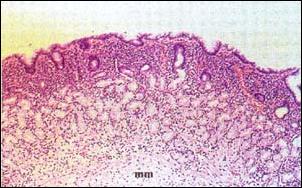
| Foto 3.- Gastritis Crónica Profunda. Infiltración linfocitica del estrato antral. También se observa Gastritis Superficial Crónica Activa (X 200) Coloración Hematoxilina y Eosina. |
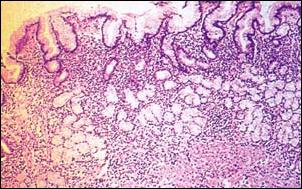
| Foto 4 .- Agregado Linfoide. Acúmulo denso de linfocitos ubicado entre glándulas antrales, reemplazándolas focalmente. Se observa un eosinófilo (flecha) en el centro del agregado. (X 300) Coloración Hematoxilina y Eosina. |
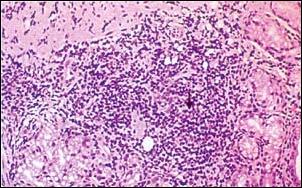
| Foto 5 .- Presencia de Linfocitos T (CD8+) en la Gastritis Profunda.Se observa con rojo las glándulas antrales parcialmente destruidas y cercano a los linfocitos citotóxicos CD8+ agresores. Muscularis mucosae (mm). (X 100) Sistema de Doble Tinción deInmunohistoquímica con anticuerpos anti-CD8+ con cromógeno DAB (marrón) para linfocitos citotóxicos vs. anti-Citoqueratina de Amplio Espectro con cromógeno Fast Red (rojo) para glándulas propis gástricas. |
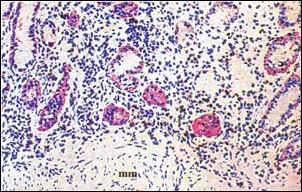
| Foto 6 .- Linfocitos T CD8+ en íntima asociación con glándulas antrales destruidas. Rezagos de Glándulas Antrales destruidas en rojo y linfocitos citotóxicos en marrón. En la parte superficial se observan glándulas antrales preservadas. (X 400) Sistema de Doble Tinción de Inmunohistoquímica con anticuerpos anti-CD8+ con cromógeno DAB (marrón) para linfocitos citotóxicos vs. anti-Citoqueratina de Amplio Espectro con cromógeno Fast Red (rojo) para glándulas propias gástricas. |
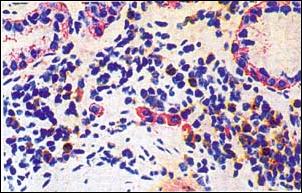
| Foto 7. - Linfocitos B (CD20+) en la zona de Gastritis Profunda, negativos en la zona superficial. (X 80) Tinción de Inmunohistoquímica con anticuerpo anti-CD20+. |
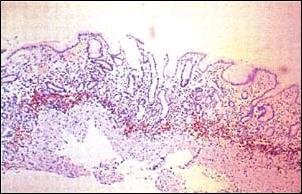
| Foto 8 .- Linfocitos B (CD20+) en Agregado Linfoide, en el que se observan glándulas gástricas en su interior. (X 300) Tinción de Inmunohistoquímica con anticuerpo anti-CD20+. |
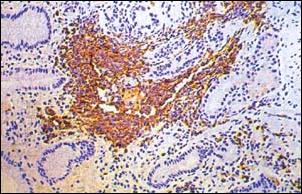
Proponemos el esquema hipotético ilustrado en las Figuras 1 a 3 para la explicación de los resultados obtenidos en el presente estudio. En la figura 1, se muestra de manera resumida, la clasificación de los linfocitos normales. En la figura 2 y 3, se ilustran los mecanismos inmunes propuestos. Ante la presencia del Helicobacter pylori, ha sido propuesto que la célula epitelial gástrica se convierte en célula presentadora de antígeno al introducir los antígenos bacterianos a su citoplasma10-11,17. Como consecuencia de ello y para poder cumplir dicha función, la célula epitelial gástrica produce un receptor del tipo MHC - II (Complejo Mayor de Histocompatibilidad tipo II) y una molécula coestimulatoria, que ayudan a presentar los antígenos bacterianos del Helicobacter pylori en su superficie8-9,11,17. Ya expuestos en la superficie de la ahora célula presentadora de antígeno, se forman un complejo con linfocitos T CD4+ o ayudadores4,7-9,11-16. Este complejo, lleva a la activación del linfocito T CD4+, el que produce citoquinas (la más importante gamma-interferón) que son responsables de la aparición de más linfocitos CD4+12-16, y a su vez de linfocitos CD8+ citotóxicos11,14 y de células B secretoras de anticuerpos7,10,12, así como también es responsable de la activación de las demás células glandulares gástricas para su conversión en células presentadoras de antígeno, creándose una cascada inflamatoria que conlleva a la destrucción de las células de las glándulas propias gástricas, ya sea por daño directo del linfocito T citotóxico CD8+, al producir perforinas en la pared de la célula que contienen antígeno de Helicobacter pylori; o indirecta, mediante la producción de anticuerpos de forma local por linfocitos B CD20+4,7,10. Esto conlleva a la destrucción de las glándulas propias gástricas y su reemplazo por tejido fibroinflamatorio y a la producción de la llamada gastritis multifocal atrófica crónica. Mayores estudios acerca de la participación de otros tipos de células como los linfocitos NK ("Natural Killer")32-33 y de otros mecanismos como el de apoptosis celular17, permitirán la mayor comprensión de la génesis de la destrucción glandular que conlleva a la atrofia gástrica.
Figura 1. Clasificación de Linfocitos.
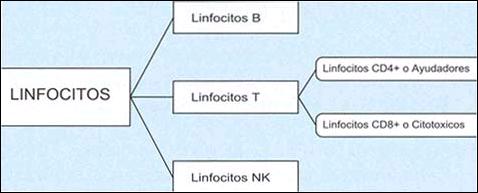
| Figura 2. Representación esquemática de la acción del Helicobacter pylori sobre el epitelio de cubierta (flecha rojo recta), Antígenos del Helicobacter pylori captados por célula glandular gástrica (flecha rojo curva) y su conservación a Célula Presentadora de Antígeno (flecha negra). |
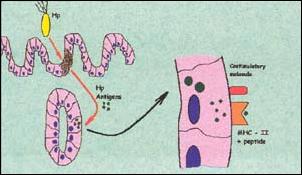
| Figura 3.- Respuesta Inmune tipo Hipersensibilidad Celular en la Gastritis Atrófica. Se muestra la presentación del antígeno del Helicobacter pylori por la Célula Presentadora de Antígeno a un Linfocito T tipo CD4+ o ayudadores, el cual se activa liberando citoquinas. Estas citoquinas son las responsables de la aparición de células tipo CD8+ o linfocitos citotóxicos y Linfocitos B en la respuesta inmune, que ulteriormente conllevan a la destrucción de las células glandulares gástricas y el desarrollo de Gastritis Atrófica. |
En nuestro medio, la gastritis por Helicobacter pylori y la GCA son comunes, incluyendo a adolescentes en hasta 33%, de pacientes sintomáticos(1). Se ha remarcado ya la importancia de la GCA como condición precancerosa. Investigaciones posteriores acerca de su génesis, permitirán intervenciones tempranas que puedan revertir la GCA y por tanto, disminuir el riesgo de cáncer gástrico.
BIBLIOGRAFÍA
1. Recavarren Arce S., Gilman R., León Barúa R. Chronic Atrophic Gastritis: Early Diagnosis in a Population Where Helicobacter pylori Infection is Frequent. Clin Infect Dis 1997; 25: 1006-12. [ Links ]
2. Dixon M., Genta R., Yardley J., Correa P. Participants in the International Workshop on Histopathology of Gastritis. Classification and Grading of Gastritis: The Updated Sydney System. Am J Surg Pathol 1994; 20(10): 1161-1181. [ Links ]
3. Hatz R., Brooks W., Krämling H., Enders G. Stomach Inmunology and Helicobacter pylori infection. Curr Opin Gastroenterol 1992; 8: 993-1001. [ Links ]
4. Ernst P., Gold B. The Disease Spectrum of Helicobacter pylori: The Immunopathogenesis of Gastroduodenal Ulcer and Gastric Cancer. Annu. Rev. Microbiol 2000; 54: 615-40. [ Links ]
5. Hui P., Chan W., Cheung P. Pathologic changes of Gastric Mucosa colonized by Helicobacter pylori. HUM PATHOL 1992; 23(5): 548-556. [ Links ]
6. Taylor K. B. Chronic Atrophic Gastritis. Actas del X Congreso Panamericano de Gastroenterología 1967; 569-571. [ Links ]
7. Wershil B. K. Immune mediators and cytokines in gastrointestinal inflammation. Curr Opin Gastroenterol 1992; 8: 975-982.8. elves P., Roitt I. The Immune System. N Engl J Med 2000; 343(1): 37-48. [ Links ]
9. Delves P., Roitt I. The Immune System. N Engl J Med 2000; 343(2): 108-117. [ Links ]
10. Faller G., Ruff S., Reiche N. Mucosal Production of Antigastric Autoantibodies in Helicobacter pylori Gastritis. Helicobacter 2000; 3(3): 129-133. [ Links ]
11. Ye G., Barrera C., Fan X., Gourley W. K. Potencial Role in CD4+T Cell Activation during Helicobacter pylori Infection. J Clin Invest 1997; 99(7): 1628-1636. [ Links ]
12. Bamford K. B., Fan X., Crowe S. Lymphocytes in the Human Gastric Mucosa During Helicobacter pylori have a T Helper Cell 1 Phenotype. Gastroenterology 1998; 114: 482-492. [ Links ]
13. Mohammadi M., Czinn S., Redline R. Helicobacter-Specific Cell-Mediated Immune Response Display a Predominant Th1 Phenotype and Promote a Delayed-Type Hypersensitivity Response in Stomachs of Mice. J Immunol 1996; 156: 4729-4738. [ Links ]
14. Fan X J, Chua A, Shani CN, McDevitt J, Keeling P W, Kelleher D. Gastric T Lymphocyte responses to Helicobacter pylori in patients with H pylori colonization. Gut 1994; 35:1379-1384 [ Links ]
15. Di Tommaso A., Xiang Z., Bugnoli M. Helicobacter pylori-Specific CD4+T-Cell Clones from Peripherial Blood and Gastric Biopsies. Infect Immun 1995; 63 (3): 1102-1106 [ Links ]
16. D'Elios M., Manghetti M., De Carli M. T Helper 1 Effector Cells Specific for Helicobacter pylori in the Gastric Antrum of Patients with Peptic Ulcer Disease. J Immunol 1997; 962-967. [ Links ]
17. Fan X., Crowe E. S., Behar S. The Effect of Class II Major Histocompatibility Complex Expression on Adherence of Helicobacter pylori and Induction of Apoptosis in Gastric Epithelial Cells: A Mechanism for T Helper Cell Type 1-mediated Damage. J Exp Med 1998; 187 (10): 1659-1669. [ Links ]
18. Haeberle H. A., Kubin M., Bamford K. B. Differential Stimulation of Interleukin-12 (IL-12) and IL-10 by Live and Killed Helicobacter pylori In Vitro and Association of IL-12 Production with Gamma Interferon-Producing T cells in the Human Gastric Mucosa. Infect Immun 1997; 65 (10): 4229-4235 [ Links ]
19. Weetman Anthony P. Chronic Autoimmune Thyroiditis. The Thyroid-A fundamental and Clinical Test 2000-First Edition; Chapter 53. [ Links ]
20. Kassan Stuart S. Sjögren Syndrome. Manual of Reumatology and Outpatient Orthopedic Disorders 2000-First Edition; Chapter 29. [ Links ]
21. Mattsson A., Tinnert A., Hamlet A. Specific Antibodies in Sera and Gastric Aspirates of Symptomatic and Asymptomatic Helicobacter pylori-Infected Subjects. Clin Diagn Lab Immunol 1998; 5(3): 288-293 [ Links ]
22. Crabtree J.E., Taylor J. D., Wyatt J. I. Mucosal IgA recognition of Helicobacter pylori 120 kDa protein, peptic ulceration, and gastric pathology. Lancet 1991; 338 (10): 332-335. [ Links ]
23. Matsukura N., Onda M., Tokunaga A. Mucosal IgA Antibody against Helicobacter pylori in Chronic Gastritis and Intestinal Metaplasia Detected by the Test-Tape Meted in Resection Specimens after Gastrectomy for Gastric Cancer. Cancer 1994; 75: 1472-1477 [ Links ]
24. Guruge J. L., Falk P. G., Lorenz R. G. Epithelial attachment alters the outcome of Helicobacter pylori infection. Proc. Natl. Acad. Sci. USA 1998; 95: 3925-3930 [ Links ]
25. Zheng P. Y, Hua J, Yeoh K G., Ho B. Association of peptic ulcer with increased expression of Lewis antigens but not cagA, iceA, and vacA in Helicobacter pylori isolates in an Asian population. Gut 2000; 47: 18-22. [ Links ]
26. Burman P., Mårdh S., Norberg L. Parietal Cell Antibodies in Pernicious Anemia Inhibit H+, K+-Adenosine Triphosphatase, the Proton Pump of the Stomach. Gastroenterol 1998; 96: 1434-1438 [ Links ]
27. Ban-Hock T., Van Driel I. Pernicious Anemia. N Engl J Med 1997; 337 (20): 1441-1448 [ Links ]
28. Genta R. M. Gastric Mucosa-Associated Lymphoid Tissue Lymphoma: Diagnosis and Therapy. Clin Perspect Gastroenterol 2000; 245-252. [ Links ]
29. Yamaguchi H., Osaki T., Masanori K. Immune Response against a Cross-Reactive Epitope on the Heat Shock Protein 60 Homologue of Helicobacter pylori. Infect Immun 2000; 68 (6): 3448-3454. [ Links ]
30. Sternberg S. Stomach-Normal Appearances. In Sternberg S. Diagnostic Surgical Pathology, 2000-Third edition, Chapter 32, 538-539. [ Links ]
31. Sternberg S. 1995. Stomach-Lymphoid Tissue. In Sternberg S. Histology for Pathologists, 1995; Chapter 27, pp. 540. [ Links ]
32. Tarkkanen J., Kosunen T. Contact of Lymphocytes with Helicobacter pylori Augments Natural Killer Cell Activity and Induces Production of Gamma Interferon. Infect Immun 1993; 61 (7): 3012-3016. [ Links ]
33. Manetti R., Parronchi P. Natural Killer Cell Stimulatory Factor (Interleukin 12 [IL-12]) Induces T Helper Type 1 (Th1)-specific Immune Responses and Inhibits the Development of IL-4-producing Th Cells. J Exp Med 1993; 177; 1199-1204. [ Links ]
34. Mahoney F. Update on Diagnosis, Management, and Prevention of Hepatitis B Virus Infection. Clin Microbiol Rev 1999; 351-356. [ Links ]
35. Kamal G. Pathologic Features of Chronic Hepatitis. A Review and Update. American J Clin Pathol 2000; 113: 40-55. [ Links ]
