










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Gastroenterología del Perú
versión impresa ISSN 1022-5129
Rev. gastroenterol. Perú v.24 n.4 Lima oct./dic. 2004
REPORTE DE CASOS
Síndrome de Peutz-Jeghers y adenocarcinoma de colon
Juan Francisco Pinto Sánchez1; Segundo Rebaza Vásquez2; Samuel Muñoz Mendoza.2; Vicente Maco Cárdenas. 3
1 Médico Gastroenterólogo. Ex-Residente UNMSM. Hospital Central de la Sanidad de la PNP
2 Médico Gastroenterólogo. Hospital Central de la Sanidad de la PNP.
3 Médico Patólogo. Hospital Central de la Sanidad de la PNP.
RESUMEN
El síndrome de Peutz-Jeghers es un desorden autosómico dominante, caracterizado por la presencia de pólipos hamartomatosos intestinales y pigmentaciones mucocutáneas características. Es un síndrome raro y se halla asociado a un alto riesgo de malignidad gastrointestinal y no gastrointestinal. Presentamos el caso de una paciente de 32 años con historia de dolor abdominal y sangrado rectal. La endoscopia digestiva alta y colonoscopia revelaron pólipos hamartomatosos y una tumoración en el colon, informada como pólipo tubular con displasia severa, por lo que fue indicado el tratamiento quirúrgico. El diagnóstico anátomo-patológico final fue un adenocarcinoma de colon.
PALABRAS CLAVE: síndrome de Peutz-Jeghers, adenocarcinoma de colon, pólipo hamartomatoso.
SUMMARY
The Peutz-Jeghers syndrome is an autosomal dominant disorder characterized by the presence of hamartomatous polyps and characteristic mucocutaneous pigmentations. It is a rare syndrome and its associated to high risk for both gastrointestinal and non-gastrointestinal malignancies. The case of a patient, 32 years old, with symptoms of abdominal pain and rectal bleeding is reported. The upper gastric endoscopy and colonoscopy showed hamartomatous polyps and a tumor in the colon. Reported as a tubular polyp with severe dysplasia, the patient underwent surgical treatment. The final anatomopathologic diagnosis was colon adenocarcinoma.
KEY WORDS: Peutz-Jeghers syndrome, colon adenocarcinoma, hamartomatous polyp.
INTRODUCCIÓN
El Síndrome de Peutz Jeghers (SPJ) es una condición rara en la cual múltiples pólipos
hamartomatosos están presentes en el tracto gastrointestinal en asociación con pigmentaciones mucocutáneas distintivas. La enfermedad fue reconocida por vez primera en 1921 por Peutz en una familia holandesa, siendo la publicación de Jeghers 28 años después 1,2 y el pedigree de ésta familia original aún continúa en estudio 3.
Es un desorden autosómico dominante y el gen relacionado ha sido mapeado en el cromosoma 19p13.3, el defecto involucra mutaciones en un gen que codifica una kinasa serina treonina 4-6 siendo el rol de ésta protein-kinasa, desconocido. Adicionalmente, en tres familias afectadas no ha sido posible establecer tal relación, lo que sugiere que existe un segundo locus 4.
El SPJ está asociado con un riesgo incrementado de malignidades gastrointestinales y no gastrointestinales 7-12, siendo la distribución de los cánceres en el tubo digestivo, similar a aquella de los pólipos hamartomatosos; y existiendo numerosos reportes de cánceres en pacientes con SPJ.
Se reporta el caso de una paciente con SPJ con historia de dolor abdominal y sangrado digestivo bajo, que fue sometida a cirugía por un cáncer de colon.
REPORTE DE CASO.
Paciente mujer de 32 años, natural y procedente de Sullana, Piura.
Antecedentes: hace aproximadamente 12 años tuvo dolor abdominal tipo cólico y sangrado rectal, los cuales eran recurrentes, por lo que recibió múltiples tratamientos para parasitosis, hasta que hace 8 años expulsó un pólipo a través del recto (de 3 cm., estudio histopatológico: adenoma tubular con displasia leve), luego de lo cual las molestias cesaron. Padre, madre y hermanos se hallan vivos y aparentemente sanos,sin presentar pigmentaciones ni síntomas gastrointestinales.
Refiere que 9 meses antes de su ingreso inicia dolor abdominal, tipo cólico, difuso; asociado a sangrado rectal, por lo cual es hospitalizada en varias oportunidades en su lugar de origen, recibiendo tratamiento para parasitosis; hace 5 meses realiza sigmoidoscopía, se halla pólipo a 25 cm del margen anal, resecándolo (no tuvo estudio histopatológico); hace 4 meses se realiza endoscopia digestiva alta, se halla pólipos duodenales. Es referida a Lima con diagnóstico de Poliposis intestinal.
Al examen clínico: buen estado general, se aprecian pigmentaciones de color marrón menores de 1 cm., distribuídas en labios, región peribucal, mucosa oral, palmas, dedos, plantas de pies y región perianal; las cuales según refiere iniciaron su aparición a los 6 años (fig. 1 y 2). El resto sin alteraciones.
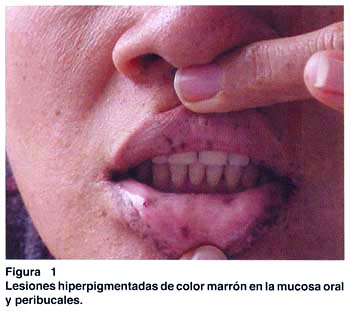
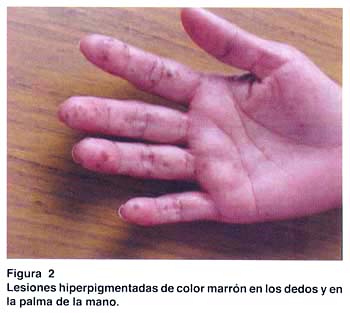
Los exámenes auxiliares excluyeron patología en tracto respiratorio alto, páncreas, hígado, vesícula biliar y vías biliares. Una ecografía transvaginal descartó patología de ovarios, útero y cérvix. Se practicó una endoscopia digestiva alta, que reveló múltiples pólipos en fondo gástrico menores de 1 cm. (estudio histopatológico: pólipos hiperplásicos) y múltiples pólipos sésiles y pedunculados en segunda y tercera porciones duodenales (pólipos hamartomatosos por histopatología); los cuales fueron resecados mediante polipectomía endoscópica (fig. 3 y 4). Una radiografía de tránsito intestinal sólo reveló dos imágenes por defecto de relleno en la segunda porción duodenal.
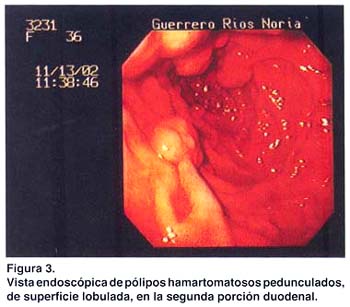
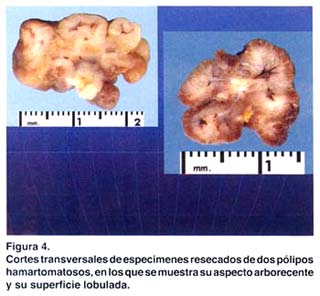
Una colonoscopía reveló pólipos colónicos; dos pólipos en colon descendente, de 8 y 10 mm. , cuya histopatología fue pólipos túbulo-vellosos. Dos en colon ascendente, de 10 y 20 mm., el estudio histopatológico informó pólipos hamartomatosos. Dos pólipos en ciego: uno de 15 mm., cuya histopatología fue pólipo hamartomatoso y otro de 25 mm., informado como pólipo hamartomatoso con hiperplasia adenomatosa focal. A nivel del ángulo esplénico del colon se observó una tumoración de aproximadamente 6-8 cm., cuyo estudio histopatológico fue informado como pólipo tubular con displasia severa.
Todos los pólipos colónicos fueron resecados endoscópicamente mediante polípectomía, luego de lo cual la paciente fue intervenida quirúrgicamente, se le realizó una colectomía segmentaria a nivel de la tumoración, resecándose aproximadamente 40 cm. de intestino grueso. Mediante exploración manual del intestino delgado se identificaron tumoraciones intraluminales, extrayéndose mediante enterotomía dos pólipos, uno de 30 mm. a 50 cm. del ángulo de Treitz y otro de 25 mm. a 50 cm. de la válvula ileocecal, ambos fueron informados como pólipos hamartomatosos en el estudio histopatológico.
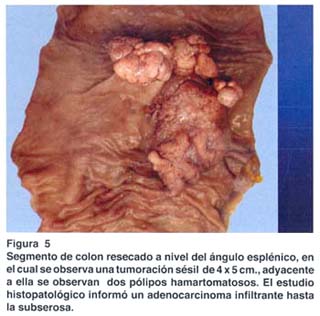
El estudio anatomopatológico de la pieza quirúrgica resecada a nivel del ángulo esplénico reveló un adenocarcinoma infiltrante hasta la subserosa, con márgenes quirúrgicos libres (fig. 5).
La paciente fue dada de alta y enviada para tratamiento quimioterápico.
DISCUSIÓN
El síndrome de Peutz-Jeghers (SPJ) es una enfermedad autosómica dominante con un alto grado de penetrancia para poliposis y pigmentación cutánea; siendo su frecuencia similar en hombres y mujeres7.
El gen responsable de éste síndrome ha sido mapeado en el cromosoma 19p13.3 y el defecto en las familias afectadas involucra mutaciones en un gen que codifica una kinasa serina treonina; las mutaciones en la línea germinal de éste gen, probablemente en combinación con defectos genéticos adquiridos del segundo alelo en células somáticas, serían responsables de las manifestaciones clínicas4-6. Otras observaciones sugieren que existe un segundo locus, probablemente localizado en el cromosoma 613.
Las manifestaciones del SPJ son dos: máculas pigmentadas mucocutáneas y múltiples pólipos hamartomatosos gastrointestinales, los cuales son benignos, pero pueden ir hacia una transformación maligna14-16. Los hamartomas se localizan mayormente en el intestino delgado (65-95 %), pero pueden también estar presentes en el colon (60 %) y estómago (50 %)7. La paciente estudiada presentó pólipos en el estómago, intestino delgado y colon.
En las series estudiadas de pacientes con SPJ, los síntomas reportados son7: obstrucción intestinal17 (por intususcepción u oclusión del lúmen por un pólipo), dolor abdominal, sangrado rectal y expulsión de un pólipo por el recto; nuestra paciente presentó como síntomas: dolor abdominal, sangrado rectal y la expulsión de un pólipo a través del recto.
Debido a que los pólipos hamartomatosos son benignos, éstos no fueron inicialmente considerados como una condición pre-maligna, sin embargo, la asociación frecuente de éste síndrome con malignidad gastrointestinal y no gastrointestinal ha conducido a la reevaluación del riesgo de cáncer en éste desorden heredado9. Se ha observado que la distribución de los cánceres gastrointestinales en éstos pacientes es similar a aquella de los pólipos hamartomatosos y que carcinomas originándose en los hamartomas han sido claramente documentados14-16.
Los pacientes con SPJ tienen un riesgo relativo 15 veces mayor para el desarrollo de cáncer que la población general18. El riesgo de cáncer gástrico, de intestino delgado y colorrectal se cree que ocurre vía cambios adenomatosos en los hamartomas, los cuales han sido encontrados en 3-6 % de hamartomas removidos de pacientes con SPJ19. La razón para el riesgo incrementado de cáncer gastrointestinal es desconocida. Otros tumores gastrointestinales que pueden ocurrir en el SPJ incluyen cáncer de páncreas y esófago 18. Nuestra paciente presentó múltiples pólipos hamartomatosos en intestino delgado y colon, y una tumoración de colon que fue inicialmente informada como pólipo tubular con displasia severa, por lo que fue operada, siendo el tumor finalmente informado como un adenocarcinoma de colon; en el mismo acto quirúrgico se extrajeron 2 pólipos intestinales, ambos mayores de 25 mm estando la cirugía indicada para la remoción de aquellos pólipos del intestino delgado que son sintomáticos o mayores de 15 mm.20.
Los pacientes con SPJ también tienen riesgo de presentar cáncer extraintestinal18: pulmón, mama, útero, ovario y tumor testicular de las células de Sertoli. En la paciente estudiada, se realizaron los exámenes necesarios para excluir cualquier otro tipo de malignidad gastrointestinal o extraintestinal , los cuales fueron todos negativos. Actualmente, la paciente se halla en seguimiento y dentro de un programa de vigilancia, dado su alto riesgo de presentar algún tipo de cáncer.
Juan Francisco Pinto Sánchez.
Jr. Bolognesi 560 Dpto. 2.
Bellavista, Callao. Lima, Perú.
Teléfono: 9927-7593. 4855864.
REFERENCIAS BIBLIOGRAFICAS.
1. PEUTZ JL. On a very remarkable case of familial polyposis of the mucous membrane of the intestinal tract and nasopharynx accompanied by peculiar pigmentations of the skin and mucous membrane. Ned Tijdschr Geneeskd 1921; 10: 134-46. [ Links ]
2. JEGHERS H, MC KUSICK VA, KATZ KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits. New Engl J Med 1949; 241: 993. [ Links ]
3. WESTERMAN A, ENTIUS M, DE BAAR E, et al. Peutz-Jeghers syndrome: 78 year follow-up of the original family. Lancet 1999; 353: 1211. [ Links ]
4. OLSCHWANG S, MARKIE D, SEAL S, et al. Peutz-Jeghers disease: most, but not all, families are compatible with linkage to 19p13.3. J Med Genet 1998; 35: 42. [ Links ]
5. JENNE D, REIMANN H, NEZU J, et al. Peutz –Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 1998; 18: 38. [ Links ]
6. HEMMINKI A, MARKIE D, TOMLINSON I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998; 391: 184. [ Links ]
7. UTSUNOMIYA J, GOCHO H, MIYANAGA T, et al. Peutz-Jeghers syndrome: Its natural course and management. Johns Hopkins Med J 1975; 136: 71. [ Links ]
8. SPIGELMAN A, MURDAY V, PHILLIPS R. Cancer and the Peutz-Jeghers syndrome. Gut 1989; 30: 1588. [ Links ]
9. GIARDIELLO F, WELSH S, HAMILTON S, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. New Engl J Med 1987; 316: 1511. [ Links ]
10. HIZAWA K, LIDA M, MATSUMOTO T, et al. Cancer in Peutz-Jeghers syndrome. Cancer 1993; 72: 2777. [ Links ]
11. BOARDMAN L, THIBODEAU S, SCHAID D, et al. Increased risk of cancer in patients with Peutz-Jeghers syndrome. Ann Intern Med 1998; 128: 896. [ Links ]
12. BOARDMAN L, PITTELKOW M, COUCH F, et al. Association of Peutz-Jeghers-like mucocutaneous pigmentation with breast and gynecologic carcinomas in women. Medicine (Baltimore) 2000; 79: 293. [ Links ]
13. MARKIE D, HUSON S, MAHER E et al. A pericentric inversion of chromosome six in a patient with Peutz-Jeghers syndrome and the use of FISH to localize the breakpoints on a genetic map. Human Genet 1998; 98:125. [ Links ]
14. PATTERSON M, KERNEN J. Epithelioid leiomyosarcoma originating in a hamartomatous polyp from a patient with Peutz-Jeghers syndrome. Gastroenterology 1985; 88: 1060-4. [ Links ]
15. MATUCHANSKY C, BABIN S, COUTROT S et al. Peutz-Jeghers syndrome with metastasizing carcinoma arising from a jejunal hamartoma. Gastroenterology 1979; 77: 1311-5. [ Links ]
16. FOLEY T, MC GARRITY T, ABT A. Peutz-Jeghers syndrome: a clinicopathologic survey of the Harrisburg family with a 49-year follow-up. Gastroenterolgy 1988; 95:1535-40. [ Links ]
17. LOZANO A, VALENCIA V, ZEVALLOS M et al. Síndrome de Peutz-Jeghers: a propósito de un reporte familiar en el hospital arzobispo Loayza. Rev gastroenterol Perú 1996; 16. 72-6. [ Links ]
18. GIARDIELLO F, BRENSINGER J, TERSMETTE a et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000; 119: 1447-53 [ Links ]
19. NARITA T, ETO T, ITO T. Peutz-Jeghers syndrome with adenomas and adenocarcinomas in colonic polyps. Am J Surg Pathol 1987; 11: 76.
20. EDWARDS D, KHOSRAVIANI K, STAFFERTON R et al. Long-term results of polyp clearance by intraoperative enteroscopy in the Peutz-Jeghers syndrome. Dis Colon Rectum 2003; 46: 48-50.
