










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Gastroenterología del Perú
versión impresa ISSN 1022-5129
Rev. gastroenterol. Perú v.26 n.3 Lima jul.-set. 2006
REPORTE DE CASOS CLÍNICOS
Hemocromatosis Hereditaria: Reporte de caso clínico y revisión de la literatura
Ricardo Prochazka1; Martín Tagle2
1 Servicio de Gastroenterología del Hospital Nacional Cayetano Heredia. Universidad Peruana Cayetano Heredia. Lima, Perú.
2 Profesor asociado de la Facultad de Medicina de la Universidad Peruana Cayetano Heredia. Médico gastroenterólogo de la Clínica Angloamericana. Lima, Perú.
RESUMEN
La hemocromatosis es una condición hereditaria que ocasiona sobrecarga progresiva de hierro como resultado de mutaciones en proteínas que regulan su absorción en el intestino. Es una enfermedad sistémica con manifestaciones variadas incluyendo cirrosis hepática, diabetes mellitus, cardiomiopatía, artropatía, y una proporción de pacientes asintomáticos. Cuando se diagnostica y se instaura tratamiento con flebotomías antes del desarrollo de daño de órganos el pronóstico es muy bueno, con una supervivencia normal, y libre de manifestaciones. La condición es común en poblaciones europeas. Reportamos un paciente peruano de ascendencia europea, asintomático, con elevación de aminotransferasas y con elevación de marcadores de hierro en quien mediante estudio genético se confirmó el diagnóstico de hemocromatosis hereditaria.
Palabras clave: hemocromatosis, aminotransferasas, hierro
SUMMARY
Hemachromatosis is a hereditary condition, producing progressive iron overload as a result of the mutation in proteins that regulate intestinal iron absorption. It is a systemic disease with several manifestations including cirrhosis, diabetes mellitus, cardiomyopathy, joint disease and a proportion of asymptomatic patients. When it is diagnosed and treatment with phlebotomies is initiated before any organ damage is developed, the prognosis is very good, with normal survival free of manifestations. This condition is common in European populations. We report the case of a Peruvian patient of European ancestry who is asymptomatic, but has high levels of aminotransferases and elevated iron markers. Genetic testing confirmed the patientís diagnosis of hereditary hemachromatosis.
Key words: hemochromatosis, aminotransferases, iron
INTRODUCCIÓN
La hemocromatosis hereditaria produce sobrecarga de hierro como resultado de mutaciones que aumentan su absorción en el intestino. El hierro se deposita produciendo manifestaciones progresivas en el hígado, varios órganos endocrinos (incluyendo páncreas, hipófisis, tiroides, y paratiroides), articulaciones, el miocardio y la piel.1 La enfermedad, que tiene el potencial de producir daño orgánico severo con expectativa de vida reducida, es frecuente en la población europea y poco común en la población latinoamericana. Reportamos un caso de hemocromatosis diagnosticado en nuestro medio y revisamos la patogenia, la clínica, y el manejo de esta interesante condición.
CASO CLÍNICO
Se presenta un varón asintomático de 44 años, peruano, hijo de padres europeos, a quien en un examen médico rutinario se le encuentra elevación de aminotransferasas y aumento de la ecogenicidad hepática, sin lesiones focales ni dilatación de vías biliares. El paciente bebe esporádicamente alcohol en cantidad moderada. No tiene antecedente de cirugías, transfusiones, tatuajes, ni conducta sexual de riesgo. No usa medicamentos, productos de medicina alternativa, tabaco, ni drogas ilícitas. Sus padres, esposa, e hija están vivos y sanos. Su examen revela buen estado nutricional con índice de masa corporal (IMC) en 24 kg/m2, sin estigmas de hepatopatía, sin edema o adenopatías, y sin anormalidades de los sistemas y aparatos. Los exámenes auxiliares sólo encontraron elevación leve de ALT, AST, y bilirrubina indirecta (Tabla 1).

Exámenes adicionales mostraron únicamente elevación de la ferritina y la saturación de transferrina (tabla 2). Se realizó un estudio genético para hemocromatosis encontrando al paciente homozigote para la mutación C282Y, confirmando el diagnóstico de hemocromatosis hereditaria.
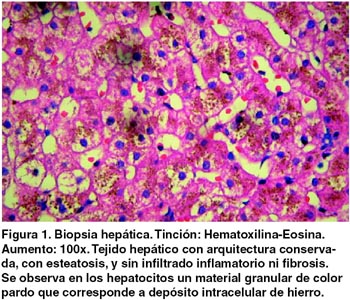
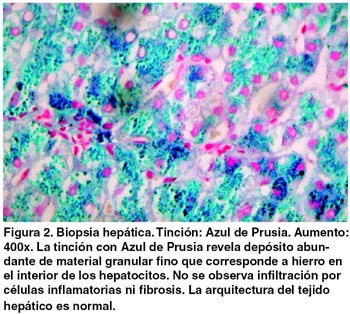
La biopsia hepática mostró abundante depósito de hierro en los hepatocitos (4+), evidente en la tinción Hematoxilina-Eosina y con Azul de Prusia, acompañado de esteatosis, sin inflamación ni fibrosis (Figuras 1 y 2).
Se inició terapia con flebotomías extrayéndose 1 unidad de sangre dos veces por semana. Luego de 40 flebotomías su ferritina bajó a 39 ng/ml, la saturación de transferrina a 18% y mantiene hemoglobina en 12 g/dl. Al haber iniciado terapia antes de desarrollar manifestaciones, su pronóstico es bueno con una expectativa de vida normal. La esposa del paciente fue negativa para mutaciones HFE, por lo que el riesgo para su hija es nulo.
DISCUSIÓN
La elevación asintomática de aminotransferasas es común en la práctica clínica. El nivel y la predominancia de ALT o AST pueden orientar en el diagnóstico (Tablas 3 y 4).2 La mayoría de casos corresponden a esteatosis y esteatohepatitis no alcohólica, enfermedad hepática alcohólica, y hepatitis viral crónica.3 En este caso, el IMC normal, la glicemia y el perfil lipídico normales, y el bajo consumo de alcohol alejan las primeras posibilidades. Se descartó hepatitis viral y autoinmune. Se midió la saturación de transferrina y la ferritina sérica con resultados marcadamente elevados, y se confirmó el diagnóstico con el hallazgo del estado homozigote para la mutación más común causante de hemocromatosis hereditaria. Este es el primer caso descrito con documentación por estudio genético en nuestro medio.


Absorción normal de hierro
La mayor parte del hierro se encuentra en los eritrocitos. Cuando éstos son destruidos el hierro pasa a los macrófagos del sistema retículo endotelial (RES) desde donde es entregado a la transferrina plasmática para ser captado nuevamente por los precursores eritrocitarios. Paralelamente el hepatocito capta hierro del plasma y lo almacena en la ferritina intracelular, liberándolo según los requerimientos del organismo. La ferritina plasmática correlaciona con el depósito de ferritina intracelular y su medición permite estimar la reserva de hierro del organismo. Además el plasma recibe hierro absorbido en el duodeno. Las únicas vías de pérdida de hierro son la descamación de células intestinales, que contienen hierro como consecuencia del proceso de absorción, y las pérdidas de sangre incluyendo la menstruación y el parto.4
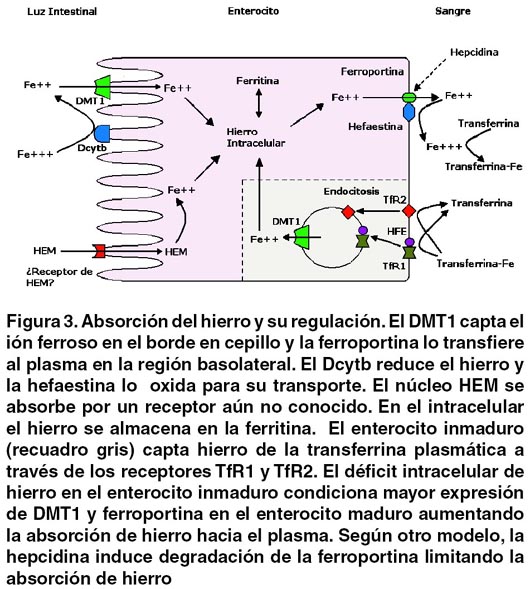
La absorción de hierro se da en el enterocito maduro de las vellosidades intestinales. En el borde en cepillo la ferrireductasa Dcytb reduce el ión férrico (Fe+++) a ferroso (Fe++) para que el transportador de metales divalentes (DMT-1) pueda trasladarlo al citoplasma. En la región basolateral la ferroportina (Fpn-1) exporta el Fe++ al extracelular y la ferroxidasa hefaestina oxida el Fe++ a Fe+++, el cual es entregado a su proteína transportadora plasmática, la transferrina.4,5,6,7
La regulación de la absorción no es completamente conocida. El enterocito inmaduro de las criptas intestinales, a través del receptor de transferrina tipo 1 (TfR1) asociado a la proteína HFE, capta hierro del plasma. El hierro intracelular del enterocito inmaduro (reflejo de la saturación de transferrina) determina la expresión de DMT-1 y Fpn-1 que tendrá enterocito maduro. Así una baja saturación de la transferrina se traduce en un nivel bajo de hierro en el enterocito inmaduro, y esto aumenta la absorción en el enterocito maduro.4,5,6,7 Según otro modelo teórico, la hepcidina, proteína plasmática sintetizada en el hepatocito, induce la degradación de la Fpn-1 disminuyendo la exportación de hierro del enterocito al plasma, limitando su absorción. Igual efecto se produce en los macrófagos, de modo que el hierro de la degradación de los eritrocitos es secuestrado en el RES ante un aumento de la hepcidina, con disminución del hierro plasmático (proceso importante en la anemia de la enfermedad crónica). La disminución de la hepcidina, por el contrario, favorece la exportación de hierro hacia el plasma desde enterocitos y macrófagos, aumentando la absorción de hierro y su disponibilidad en plasma, y disminuyendo el depósito en el RES.4,5,6,7,8 La producción de hepcidina es regulada por varios factores incluyendo anemia, hipoxia, inflamación, hierro intracelular hepático, la proteína HFE, el receptor de transferrina 2 (TfR2), y la hemojuvelina.8,9 (Figura 3)
Patogenesis de la hemocromatosis hereditaria
Alrededor del 90% de los pacientes tienen hemocromatosis tipo 1, de herencia recesiva, originada por mutaciones en la proteína HFE. Las mutaciones más comunes son la C282Y y la H63D. 80% de los pacientes son homozigotes C282Y y casi todos los demás son heterozigotes compuestos C282Y/H63D.1,5,6,7,10 Alrededor de 0.5% de la población del norte de Europa tiene el genotipo C282Y/C282Y.7,10 La mutación aumenta la absorción de hierro y la saturación de transferrina aumenta. La liberación del hierro de los macrófagos es normal. El depósito se da en hepatocitos y otras células parenquimales, y el nivel de ferritina plasmática aumenta. El genotipo no basta para desarrollar la enfermedad, y existen factores modificadores, ambientales como el consumo de alcohol, y genéticos.10
En la hemocromatosis tipo 2 o juvenil la sobrecarga de hierro causa manifestaciones en la segunda o tercera década de la vida. Tiene dos variantes: 2a, por mutaciones de la hemojuvelina, y 2b, por mutaciones de la hepcidina5,11. La hemocromatosis tipo 3, es causada por mutaciones del TfR2. La hemocromatosis tipo 4 es causada por mutaciones en la ferroportina que impiden la liberación de hierro desde el RES y aumentan secundariamente la absorción de hierro. Su fenotipo es diferente: tendencia a la anemia, acumulación de hierro en macrófagos (con menor potencial de daño orgánico), y pobre tolerancia a la terapia con flebotomías.5,11
Manifestaciones de la hemocromatosis hereditaria
La hemocromatosis es una enfermedad sistémica con manifestaciones variadas y una proporción de pacientes asintomáticos. La tríada de hiperpigmentación, hepatopatía, y diabetes sólo ocurre en 7% de los pacientes.12,13 Las manifestaciones aparecen a edades entre 40 y 60 años. 5,6,11 Alrededor del 75% de los varones y 40% de las mujeres con genotipo de hemocromatosis tipo 1 tienen marcadores de hierro aumentados, pero sólo 1.2% desarrollan síntomas.10,14,15 Las mujeres tienen menor riesgo al estar protegidas por la hemorragia menstrual.16
En homozigotes C282Y identificados en población general se encuentra enfermedad hepática, AST elevada, y colágeno tipo IV plasmático elevado (indicador de fibrosis hepática) con mayor frecuencia que en individuos sin mutaciones HFE.13,15,17,18,19 En poblaciones clínicas con hemocromatosis 22% tienen cirrosis.12 Todos los homozigotes C282Y con fibrosis importante o cirrosis tienen más de 40 años.20 Hepatomegalia, AST elevada, y ferritina superior a 1000 ng/ml son predictores de fibrosis severa o cirrosis.21 Por ello los homozigotes C282Y con esas características deben ser sometidos a una biopsia hepática.22 El riesgo de cáncer de hígado y vías biliares es 200 veces mayor que en la población general, incluso años después de la remoción del exceso de hierro.23,24,25
La prevalencia de cardiopatía en pacientes con hemocromatosis varía entre 7.8% y 35%.12,16,25 Las manifestaciones son cambios electrocardiográficos, fibrilación auricular, síndrome del seno enfermo, dilatación de cámaras, y disfunción sistólica. 23,26 Clínicamente se producen arritmias o insuficiencia cardíaca.12,16,25 La insuficiencia cardíaca puede ser rápidamente fatal y la respuesta a la terapia es variable.12,16,23
La acumulación de hierro reduce la producción de insulina y disminuye la sensibilidad a la misma.23 Se describe diabetes mellitus entre el 14 y el 65% de los pacientes con hemocromatosis.12,16 La remoción del hierro no elimina la dependencia de insulina pero disminuye la dosis requerida25. Otros problemas endocrinológicos en pacientes con hemocromatosis son hipotiroidismo e hipogonadismo, que se manifiesta como impotencia, amenorrea y pérdida de la libido23.
Entre 40 y 75% de pacientes con hemocromatosis tienen artropatía. Se comprometen las articulaciones metacarpofalángicas 2° y 3° con osteofitos, estrechamiento del espacio articular, osteoporosis subcondral, lesiones quísticas y condrocalcinosis. El compromiso articular no responde a la remoción de hierro.27,28
La expectativa de vida antes del desarrollo de cirrosis o diabetes es igual que la de la población general. Los pacientes con cirrosis o diabetes tienen mortalidad aumentada. Las causas de muerte son: cáncer de hígado y vías biliares, cirrosis, cardiomiopatía, y diabetes. El pronóstico depende del nivel de sobrecarga de hierro y el momento en que ésta se interrumpe con la terapia.25
DIAGNÓSTICO
Debe considerarse el diagnóstico en pacientes con clínica sugerente, elevación asintomática de aminotransferasas, familiares de personas previamente diagnosticadas, y personas con marcadores de hierro incrementados.22
El primer paso es la medición de la ferritina sérica y la saturación de transferrina en ayunas.22,29 Si hay elevación de la saturación de transferrina el diagnóstico debe confirmarse determinando la presencia de la mutación C282Y. La elevación aislada de la ferritina es poco específica ya que puede elevarse en condiciones inflamatorias y neoplásicas.29
Si la ferritina es mayor a 1000 ng/ml, el paciente tiene más de 40 años, hay elevación de aminotransferasas, o evidencia clínica de hepatopatía, se debe realizar una biopsia hepática para buscar fibrosis o cirrosis, lo cual tiene importancia pronóstica.22 La biopsia también puede considerarse en presencia de alcoholismo, otras manifestaciones de hemocromatosis como diabetes mellitus, o en heterozigotes compuestos C282Y/H63D22.
Terapia
El hierro se remueve mediante flebotomías. Estas se inician cuando la ferritina llega a 200 ng/ml en mujeres ó 300 ng/ml en varones, antes del desarrollo de síntomas.16,22,23 Se remueven 1 ó 2 unidades de sangre por semana. Antes de cada flebotomía se mide el hematocrito para no causar caída superior a 20% del hematocrito inicial. La ferritina se mide cada 10 ó 12 flebotomías. El objetivo es una ferritina entre 25 y 50 ng/ml. Luego se continúa con 3 ó 4 flebotomías al año y medición de ferritina cada 1 ó 2 años.16,22
La enfermedad avanzada debería ser tratada ya que algunas manifestaciones pueden mitigarse con la terapia, incluyendo la severidad de la hipertensión portal.22,23,25,30 La progresión de cirrosis es indicación de transplante hepático.22 Todos los cirróticos deben tener vigilancia de por vida por riesgo de hepatocarcinoma con alfa feto proteína cada 4-6 meses.22,29
Despistaje
Entre los familiares de pacientes con hemocromatosis, 69 a 96% de los homozigotes C282Y tendrán sobrecarga de hierro, y 10-38% desarrollarán enfermedad. La mayor probabilidad de ser homozigotes corresponde a los hermanos del paciente y algo menor es el riesgo de los hijos.31 Para el despistaje en familiares, después de dar información adecuada, se propone el estudio genético. La edad óptima para la evaluación es de 20 a 40 años. En los hermanos adultos se realiza directamente el estudio genético. En los hijos la estrategia es diferente: se evalúa al cónyuge. Si éste resulta portador mutaciones HFE, a todos los hijos se les propone el estudio al llegar a la adultez. A los homozigotes o heterozigotes compuestos se les miden los indicadores de hierro cada 5 a 10 años.31 En los niños deben tenerse en cuenta aspectos éticos relacionados a la posibilidad de discriminación futura por parte de compañías de seguros y empleadores.31
CONCLUSIÓN
Se describe el primer caso en nuestro medio de hemocromatosis hereditaria con documentación por biopsia hepática con tinción de Azul de Prusia y estudios genéticos confirmando el estadío de homozigote para la mutación C282Y, en un varón asintomático, durante investigación de aminotransferasas elevadas. La evaluación de todo paciente con dicho problema frecuente en la práctica médica debe incluir en forma rutinaria la medición de Ferritina, Hierro Sérico y Saturación de Transferrina. La hemocromatosis es una condición hereditaria que ocasiona sobrecarga de hierro cuyas manifestaciones abarcan varios órganos y sistemas. El tratamiento, que consiste en la remoción del exceso de hierro mediante flebotomías, evita las consecuencias de la hemocromatosis y permite una supervivencia normal.
REFERENCIAS BIBLIOGRÁFICAS
1. PIETRANGELO A. Haemochromatosis. Gut 2003;52(Suppl II): ii23–ii30. [ Links ]
2. AMERICAN GASTROENTEROLOGICAL ASSOCIATION CLINICAL PRACTICE COMMITTEE. AGA Technical Review on the Evaluation of Liver Chemistry Tests. Gastroenterology 2002;123:1387-1394.
3. CLARK JM, BRANCATI FL, DIEHL AM. The Prevalence and Etiology of Elevated Aminotransferase Levels in the United States. Am J Gastroenterol 2003;98:960-967.
4. PIETRANGELO A. Physiology of iron transport and the hemochromatosis gene. Am J Physiol Gastrointest Liver Physiol 2002;282: G403–G414.
5. SIAH CW, TRINDER D, OLYNYK JK. Iron overload. Clin Chim Acta 2005;358:24–36.
6. PAPANIKOLAU G, PANTOPOULOS K. Iron metabolism and toxicity. Toxicol Appl Pharmacol 2005;202:199– 211.
7. WORWOOD M. Inherited iron loading: genetic testing in diagnosis and management. Blood Rev 2005;19:69–88.
8. VERGA FALZACAPPA MV, MUCKENTHALER MU. Hepcidin: Iron-hormone and anti-microbial peptide. Gene 2005;364:37–44.
9. GANZ T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003;102:783-788.
10. WAALEN J, NORDESTGAARD BG, BEUTLER E. The penetrance of hereditary hemochromatosis. Best Pract Res Clin Haematol 2005;18:203–220.
11. PIETRANGELO A. Hereditary Hemochromatosis - A new look to an old disease. N Engl J Med 2004;350:2383-97.
12. ADAMS PC, DEUGNIER Y, MOIRAND R, BRISSOT P. The Relationship Between Iron Overload, Clinical Symptoms, and Age in 410 Patients With Genetic Hemochromatosis. Hepatology 1997;25:162-166.
13. ASBERG A, HVEEM K, THORSTENSEN K, ELLEKJAER E, KANNELONNING K, FJOSNE U et al. Screening for Hemochromatosis: High Prevalence and Low Morbidity in an Unselected Population of 65,238 Persons. Scand J Gastroenterol 2001;36:1108 –1115.
14. MC CUNE CA, AL-JADER LN, MAY A, HAYES SL, JACKSON HA, WOEWOOD M. Hereditary haemochromatosis: only 1% of adult HFE C282Y homozygotes in South Wales have a clinical diagnosis of iron overload. Hum Genet 2002;111:538–543.
15. BEUTLER E, FELITTI VJ, KOZIOL JA, HO NJ, GELBART T. Penetrance of 845G A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet 2002;359:211–18.
16. ONEIL J, POWELL L. Clinical Aspects of Hemocromatosis. Semin Liver Dis 2005;25:381-391.
17. WAALEN et al. Penetrance of Hemochromatosis. Blood Cells Mol Dis 2002;29:418–432.
18. ADAMS PC, REBOUSSIN DM, BARTON JC, MCLAREN CE, ECKFELDT JH, MCLAREN GD et al. Hemochromatosis and Iron- Overload Screening in a Racially Diverse Population. N Engl J Med 2005;352:1769-78.
19. OLYNYK JK, CULLEN DJ, AQUILIA S, ROSSI E, SUMMERVILLE L, POWELL L. A Population-Based Study of the Clinical Expression of the Hemochromatosis Gene. N Engl J Med 1999;341:718-724.
20. BACON BR, OLYNYK JK, BRUNT EM, BRITTON RS, WOLFF RK. HFE Genotype in Patients with Hemochromatosis and Other Liver Diseases. Ann Intern Med 1999;130:953-962.
21. GUYADER D, JACQUELINET C, MOIRAND R, TURLIN B, MENDLER MH, CHAPERON J, DAVID V ET AL. Non-invasive prediction of fibrosis in C282Y homozygous hemocromatosis. Gastroenterology 1998; 115: 929-936.
22. TAVILL AS. Diagnosis and Management of Hemochromatosis. Hepatology 2001;33:1321-1328.
23. YEN ET AL. Revisiting Hereditary Hemocromatosis. Am J Med 2006;119:391-399.
24. BRADBEAR RA, BAIN C, SISKIND V, SCHOFIELD FD, WEBB S, AXELSEN EM et al. Cohort study of internal malignancy in genetic hemochromatosis and other chronic nonalcoholic liver diseases. J Natl Cancer Inst 1985;75:81-4.
25. NIEDERAU C, FISCHER R, PURSCHEL A, STREMMEL W, HAUSSINGER D, STROHMEYER G. Long-term Survival in Patients With Hereditary Hemochromatosis. Gastroenterology 1996;110:1107–1119.
26. OLSON LJ, BALDUS WP, TAJIK AJ. Echocardiographic features of idiopathic hemochromatosis. Am J Cardiol 1987; 60: 885-889.
27. VON KEMPIS J. Arthropathy in hereditary hemochromatosis. Curr Opin Rheumatol 2001;13:80–83.
28. VAIOPOULOS G, PAPANIKOLAU G, POLITOU M, JIBREEL I, SAKELLAROPOULOS N, LOUKOPOULOS D. Arthropathy in Juvenile Hemochromatosis. Arthritis Rheum 2003;48:227–230.
29. DOOLEY JS. Diagnosis and management of genetic haemochromatosis. Best Pract Res Clin Haematol 2002;15:277-293.
30. FRACANZANI AL, FARGION S, ROMANO R, CONTE D, PIPERNO A, DALBA R et al. Portal hypertension and iron depletion in patients with genetic hemochromatosis. Hepatology 22:1127-1131.
31. OMBIGA J, ADAMS LA, TANG K, TRINDER D, OLYNYK JK. Screening for HFE and iron overload. Semin Liver Dis 2005;25:402- 410.
