










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Gastroenterología del Perú
versión impresa ISSN 1022-5129
Rev. gastroenterol. Perú v.26 n.4 Lima oct./dic. 2006
REPORTE DE CASOS
Dolor Abdominal Agudo debido a Infarto Esplénico en un paciente con Enfermedad Heterocigota de Células Falciformes expuesto a la altura
Acute Abdominal Pain due to Splenic Infarction in a patient with Heterozygous Scikle Cell disease exposed to high altitude
Edgar Ruiz Semba1, Jorge Garavito Rentería1, Jorge Jiménez Bustamante1, Ronal Arteaga Caro1, José Luis García Del Aguila1, Vannya Chávez Gil1
1 Servicio de Medicina Interna, Hospital Arzobispo Loayza. Líma- Perú
RESÚMEN
La hemoglobinopatía S, depranocitosis ó enfermedad de células falciformes constituye la hemoglobinopatía más frecuente en el mundo. En su forma heterocigota (Sickle Cell Trait) afecta el 8% de la población negra de los EUA y al 25% de la población negra africana, y en menor frecuencia en la zona del mediterráneo, India, medio oriente y américa latina. La alteración básica es la sustitución del ácido glutámico de la posición 6 de la cadena beta de Globina por Valina, la cual polimeriza a baja tensión de oxígeno; distorsionando la estructura del hematíe, aumentando la viscosidad sanguínea, bloqueando la circulación arterial capilar de diferentes áreas del organismo produciendo así microinfartos. Aunque el Infarto esplénico es raro, es reconocido como una dramática complicación de la enfermedad heterocigota de células falciformes (Sickle Cell Trait).
Presentamos el caso de un paciente varón de 21 años de edad, mestizo, que cursa con un cuadro agudo de dolor abdominal posterior al arribo a la ciudad minera de Casapalca (ubicado en la Sierra del Perú; a 4200 msnm) por causas laborales y es derivado a nuestro Hospital en Líma para estudio. Presentamos el caso por ser una inusual causa de dolor abdominal agudo y por ser esta entidad poco frecuente en nuestro medio y con escasas publicaciones al respecto.
Palabras Claves: Enfermedad heterocigota de células falciformes, Infarto esplénico
SUMMARY
Hemoglobinopathy S, Depranocytosis or Sickle Cell Disease is the most common hemoglobinopathy in the world. In its heterozygous form (Sickle Cell Trait), it affects 8% of the black population in the U.S. and 25% of the black population in Africa, and is found less frequently in the Mediterranean area, India, Middle East and Latin America. The basic alteration is a substitution of glutamic acid by valin in the sixth position of the beta globin chain, which causes polymerization at low oxygen tension thereby distorting the structure of erythrocytes and increasing blood viscosity, which, in turn, generates obstructions of the capillary arterial blood flow to different areas of the body thus causing microinfarctions. Although Splenic Infarction is rare, it is recognized as a serious complication of Heterozygous Sickle Cell Disease (Sickle Cell Trait).
We present the case of a 21 year-old mestizo male patient who came in with an acute case of abdominal pain after arriving to work in the Casapalca mining city (located in the Peruvian Andes at 4200 m.a.s.l.) and was referred to our Hospital in Lima for exams. We present the case because it is an unusual cause of acute abdominal pain, and because this condition is rare in Peru and there are few publications about it.
KEY WORDS: Heterozygous Sickle Cell Disease, Splenic Infarction
INTRODUCCIÓN
La Depranocitosis o falciformismo es una condi-ción que en su forma heterocigota puede afectar al 8 % de la población negra americana y al 25 % de la población negra africana, reportándose en menor frecuencia en zonas del mediterráneo, India, Medio Oriente y América Latina.(1.16)
La Prevalencia tiene correlación con zonas geográficas endémicas de malaria y representa un mecanismo de protección a la infestación por el Plasmodium falciparum.
La alteración básica es la sustitución del ácido glutámico por valina en la posición 6 de la cadena beta globina. El resultado es un tetrámero de hemoglobina Alfa 2/Beta S2, pobremente soluble cuando es desoxigenado, lo cual produce la polimerización de la hemoglobina. Este especto es esencial en los fenómenos venooclusivos de la enfermedad que puede ocasionar disfunción orgánica e inclusive ser causante de muerte del individuo
CASO CLÍNICO
Paciente varón de 21 años de edad, mestizo, natural de Sullana (ubicada en la costa norte del Perú), fumador crónico, con antecedentes de estreñimiento crónico. Horas después de su arribo a la ciudad de Casapalca (localidad minera ubicada a 4200 msnm en la sierra del Perú) presenta un cuadro agudo de dolor abdominal tipo cólico de progresiva intensidad, localizado en el cuadrante superior izquierdo, asociado a vómitos de contenido bilioso, por lo cual acude a un centro de salud zonal donde le administran antibióticos y es referido a nuestro hospital para estudio. Al ingreso el paciente se encontraba hemodinámicamente estable, orientado, al exa-men físico destacaba la palpación del bazo con espacio de Traubbe ocupado. Se realiza una ecografía abdominal la cual informa Esplenomegalia con cambios en su ecoestructura por lo cual se solicita TAC abdominal observándose áreas hipodensas, no captadoras de contraste en el interior del Bazo el cual media 104 x 45mm en su diámetro transverso y anteroposterior, Luego se le solicita Eco Doppler donde se confi rman 4 áreas con ausencia de flujo compatibles con zonas de Infarto Esplénico.
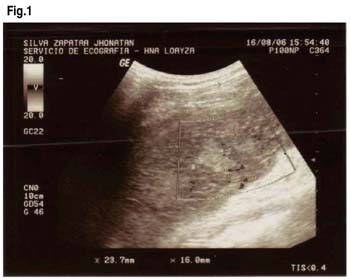
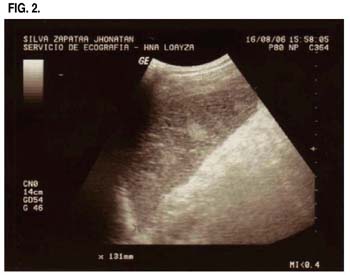
Con el diagnóstico planteado de Infarto Esplénico se realizan los siguientes exámenes, destacando: Leucocitos: 18,300 ( B: 0%, A: 3,E: 1%,S: 82%,L: 10%); Hb: 14gr/dl; Hto:41% ; Frotis de Sangre Periférica con Hipocromía y Anisocitosis; plaquetas normales; Amilasa: 77 U/I; Sedimento Urinario con 5-10 leucocitos por campo, cilindros granulosos, filamentos mucoides 1+; Glucosa: 82mg/dl; Urea:31mg/dl; T.P. 14 seg, INR: 1.16 ,85.5%; Grupo: O-Rh+; Perfi l Hepático dentro de límites normales; VIH : Negativo, HCV: negativo HBsAg: negativo; Crioaglutininas: Negativo. Se solicitó entonces Electroforesis de Hemoglobina: Determinación de Hb A1 : 59% (VN: 94-99%) Hb A2: 2.3% (VN: 1,0-3,5%) Hb Fetal: 1.7%, Hb S: 37.0% (VN:0%), conclusión: Heterocigote de Hemoglobina AS. Se le solicita un hemograma de control a los 5 días con disminución del número de leucocitos a 10,600(A:0%) Hb:12.4 gr/dl y en el Sedimento Urinario con presencia de algunos hematíes.
El paciente experimenta una notable mejoría clínica con tratamiento sintomático conservador, y sale de alta a los 8 días de su ingreso en el pabellón.
DISCUSIÓN
Se reporta el caso de un paciente varón de 21 años de edad, mestizo, natural y procedente de Sullana, Piura, caracterizado por la presencia de dolor abdominal de presentación aguda, localizado en el cuadrante superior izquierdo luego de arri-bar a la ciudad de Casapalca (ciudad minera situada en la sierra central del Perú a 4200 msnm), quién fue transferido al servicio de emergencia del Hospital Nacional Arzobispo Loayza, debido a la persistencia y severidad del cuadro doloroso abdominal.
El síndrome doloroso abdominal es un problema médico muy frecuente observado en las salas de emergencia de los hospitales, de causa muy variada. La localización del dolor es una guía de relevancia clínica que permite una aproximación diagnóstica en función al órgano u órganos subyacentes. En el caso descrito se postuló la posibilidad diagnóstica de una pancreatitis aguda sin embargo, no se encontró correlación clínica: dolor en epigastrio y/o cuadrante superior izquierdo asociado a nauseas e irradiado a espalda, ni enzimático: amilasa dentro de limites normales; la otra hipótesis planteada de una enfermedad diverticular complicada en la cual la presentación clínica de la diverticulitis depende de la extensión de la perforación y comúnmente los pacientes tienen microabcesos de pared asociado a dolor abdominal, fiebre y presencia de sensibilidad o masa en el cuadrante inferior izquierdo, fue descartada. En este punto un estudio ecográfico reveló la presencia de áreas hipoecogénicas en bazo sugestivas de un infarto esplénico.
El paciente fue transferido al servicio de medicina en donde se realizaron estudios de imágenes consistentes en Tomografía Abdominal y también de Ecografía Doppler las que confirmaron la presencia de imágenes compatible con múltiples infartos esplénicos.
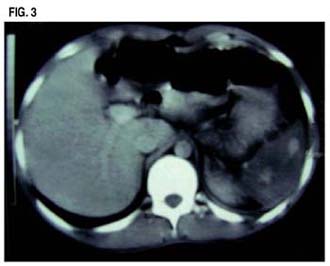
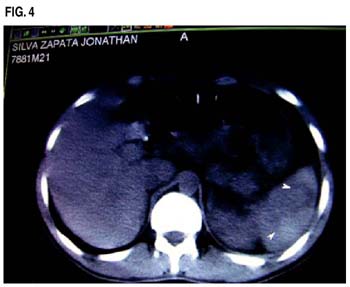
El infarto esplénico es una condición poco frecuente asociado a dolor abdominal. Se han descrito principalmente patologías cardiovasculares: Endocarditis y fibrilación auricular como causas más importantes de esta condición. Otras condiciones asociadas a infarto esplénico son: Síndrome antifosfolipídico, Lupus eritematoso sistémico, crioglobulinemia y las hemoglobinopatías. (6,13,14)
La evaluación del paciente no reveló presencia de patología cardiaca ni enfermedad autoinmune asociada e estado de hipercoagulabilidad. Sin embargo al considerar el antecedente epidemiológico de su procedencia (Piura, ciudad costera situada en el norte del Perú y endémica de malaria) y la presencia de dolor abdominal desencadenado por la exposición a la altura se postuló la posibilidad diagnóstica de una hemoglobinopatía. El estudio de electroforesis de hemoglobina, confirmó la presencia de una depranocitosis o falciformismo (Sickle cell) en su variedad heterocigota de células falciformes (Sickle cell Trait).
La depranocitosis es una hemoglobinapatía debido a la presencia de hemoglobina anormal cuyas manifestaciones clínicas esenciales son: Hemólisis y fenómenos venooclusivos. Estos últimos se manifi estan por episodios dolorosos recurrentes (Crisis Sickle cell) que puede afectar a una variedad de órganos ocasionando muchas veces disfunción en incluso la muerte del paciente. (5,6,8,9)
Se describen varios síndromes depranocíticos en función del porcentaje de hemoglobina A, S y F presente.
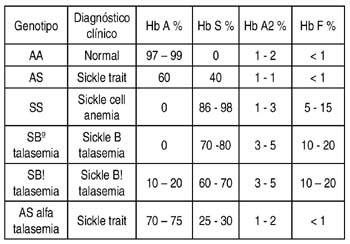
La variante Heterocigota de células falciformes (Sickle cell trait) fundamentalmente se caracteriza por la presencia de fenómenos veno-oclusivos asociados a la exposición de grandes alturas (hipoxemia) y ejercicios extenuantes, siendo hematológicamente normal. Se ha descrito defectos de la función tubular renal con incapacidad para concentrar orina y asociado a hematuria. (1,3,4,6,15)
El caso descrito corresponde a un síndrome doloroso abdominal por infarto esplénico asociado a enfermedad heterocigota de células falciformes, condición que deberá tenerse en cuenta en los cuadros de dolor abdominal agudo asociado a la exposición de grandes alturas.
El tratamiento fue conservador consistente en hidratación, analgesia y adecuada oxigenación, La evolución fue favorable estando en condiciones de alta a los 8 días de su hospitalización del Servicio de Medicina
BIBLIOGRAFIA
1. MARTIN TW, WEISSMAN IM, Exercise and Hipoxya increase sickling in venous blood from an exercising limb in individuals with sickle cell trait. Am J Med 1989, 87: 48-56. [ Links ]
2. FRISANCHO, O; FRISANCHO, D; MOLINA, CS; HEREDIA, J; LEÓN, A. Infarto de bazo y altura. Rev. gastroenterol. Perú 1984; 4:15-25. [ Links ]
3. LANE P. A, GITHENS J. H. Splenic syndrome at mountain altitudes in sickle cell trait. Its occurrence in nonblack persons. JAMA 1985; 253. [ Links ]
4. KING D. T., LINDSTROM R. R., STATE D., HIROSE F. M. AND SCHWARTZ A. Unusual cause of acute abdomen. Sickle cell trait and nonhypoxic splenic infarction. JAMA 1977; 238. [ Links ]
5. CLASTER S., VICHINSKY E. Managing sickle cell disease. BMJ, 327: 1151-55 [ Links ]
6. BUCHANAN GR., DEBAUN M., QUINN C., STEINBERG M. Sickle Cell Disease. American Society of Hematology. Hematology 2004; 35-47. [ Links ]
7. MORRIS C., RUCKNAGEL D., AND JOINER C. Deoxygenation - Induced Changes in Sickle Cell-Sickle Cell Adhesion. Blood. 1993; 81: 3138-3145 [ Links ]
8. REGINALD P. PUCH, THOMAS V. MONICAL AND VIRGINIA MINNICH. Sickle Cell Anemia with Two Adult Hemoglobins S and Hb GPhiladelphia/S. BLOOD 1964; 23: 206-215. [ Links ]
9. STEINBERG M., Management of sickle cell disease. NEJM 1999; 340:1021-30 [ Links ]
10. WALTERS M. Stem Cell Therapy for Sickle Cell Disease: Transplantation and Gene Therapy. American Society of Hematology. Hematology 2005; 66-73 [ Links ]
11. LOTTENBERG R., HASSELL K. An Evidence-Based Approach to the Treatment of Adults with Sickle Cell Disease. American Society of Hematology. Hematology 2005; 58-65. [ Links ]
12. RAMÍREZ J. LIZAMA O., MARTÍNEZ J., JHONG M., SALAZAR L. Reporte de un probable caso de Hemoglobina S / Talasemia Beta. Rev Med Hered 2004; 15: 173-178. [ Links ]
13. CHARLES PETERSON, MD, MBA. Sickle cell anemia, Nacional Hearth, Lung and blood Institute web site, May 2006. [ Links ]
14. FRANKLIN H. EPSTEIN, M.D., Editor. Pathogenesis and Treatment
of Sickle Cell Disease. NEJM 1994; 11:762-769.
15. JHON B. WEST, MD., PhD. The Physiologic Basis of High-Altitude Diseases. Ann Intern Med. 2004; 141:789-800. [ Links ]
16. SAMAAD MALIK, Acute Splenic Infarction, CMAJ. 2006; 175; 247 [ Links ]
