










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Gastroenterología del Perú
versión impresa ISSN 1022-5129
Rev. gastroenterol. Perú v.28 n.2 Lima abr./jun. 2008
REPORTE DE CASOS
Colestasis intrahepática benigna recurrente y su progresión a colestasis intrahepática familiar progresiva
Recurrent benign intrahepatic cholestasis and their progressión to familiar progressive intrahepatic cholestasis
Marco Alburquerque Miranda1, Gloria Vargas Cárdenas2, Zenaida Lozano Miranda3, Tania Reyes Mugruza1, Walter Li Torres4, Mario Valdivia Roldán2
1 Médico Residente del Servicio de Gastroenterología del Hospital Nacional Arzobispo Loayza. Lima – Perú.
2 Médico Asistente del Servicio de Gastroenterología del Hospital Nacional Arzobispo Loayza. Lima – Perú.
3 Médico Asistente del Servicio de Anatomía Patológica del Hospital Nacional Arzobispo Loayza. Lima – Perú.
4 Interno de Medicina del Hospital Nacional Arzobispo Loayza. Lima – Perú.
RESUMEN
La Colestasis Intrahepatica Benigna Recurrente (CIBR) es una rara forma de colestasis intrahepatica caracterizada por episodios recurrentes y autolimitados de ictericia y prurito intensos. Clásicamente su evolución natural es benigna, sin progresión a fibrosis o insuficiencia hepática; sin embargo, últimamente se han reportado casos que progresan a Colestasis Intrahepatica Familiar Progresiva (CIFP), ésta última caracterizada por insuficiencia hepática y cirrosis. Presentamos el caso de un paciente varón de 32 años que acude al Servicio de Gastroenterología del Hospital Nacional Arzobispo Loayza, por ictericia y prurito. Lo reportamos por lo infrecuente de su presentación y por ser una entidad que debemos tener en cuenta en el diagnóstico diferencial de enfermedades hepáticas colestásicas.
PALABRAS CLAVES: Colestasis Intrahepatica Benigna Recurrente (CIBR). Colestasis Intrahepatica Familiar Progresiva (CIFP). Enfermedades hepáticas colestásicas.
ABSTRACT
Benign recurrent intrahepatic cholestasis (BRIC) is a rare form of intrahepatic cholestasis characterized by repeated self-limited episodes of severe pruritus and jaundice. Classically its natural evolution is benign, without progress to fibrosis or hepatic insufficiency; although, lastly were reported cases which progress to Progressive familial intrahepatic cholestasis (PFIC). This disease is characterized by progressive hepatic insufficiency and cirrhosis. We present the case of a 32 years old male patient who went to Gastroenterology Service of Arzobispo Loayza National Hospital by pruritus and jaundice. We reported this case for its infrequent presentation and because is an entity which should be considered within differential diagnosis of hepatic cholestasis diseases.
KEY WORDS: Benign recurrent intrahepatic cholestasis (BRIC). Progressive familial intrahepatic cholestasis (PFIC). Hepatic cholestasis diseases
INTRODUCCIÓN
La colestasis es un término genérico que representa una alteración del fl ujo biliar y se caracteriza desde el punto de vista bioquímico por mayor elevación de los niveles séricos de fosfatasa alcalina o de bilirrubina conjugada con respecto a los niveles de aminotransferasas( 1). En este trastorno los síntomas y signos clínicos derivan de la acumulación de sustancias habitualmente excretadas en la bilis que se depositan en el hígado, sangre y otros tejidos; y de la malabsorción de grasas y vitaminas liposolubles por disminución de ácidos biliares en el intestino delgado(2).
Se distingue entre colestasis intra y extrahepática, según donde se localice la alteración del fl ujo biliar. En el primer caso la localización puede estar situada en cualquier punto entre el citoplasma de los hepatocitos y los conductos biliares de mediano calibre (100-400 μm). En las colestasis extrahepáticas la lesión obstructiva se encuentra en los conductos biliares grandes que en la mayoría de los casos es debida a litiasis o a tumores pancreatobiliares, incluyendo ampulomas o adenopatías hiliares(3).
Las causas de colestasis intrahepatica son diversas y van desde las enfermedades autoinmunes: Cirrosis biliar primaria y Colangitis esclerosante primaria, como las mas comunes; hasta los trastornos hereditarios del fl ujo biliar como la Colestasis Intrahepatica recurrente benigna y Colestasis Intrahepatica familiar progresiva, siendo estas últimas las formas más raras(4,5).
El presente caso se trata de un paciente con diagnostico de Colestasis Intrahepatica familiar progresiva tipo 1 que fue diagnosticado en el Servicio de Gastroenterologia y Medicina del Hospital Nacional Arzobispo Loayza y lo reportamos por lo infrecuente de su presentación y por ser una hepatopatia colestásica que debemos tener en cuenta en el diagnostico diferencial de enfermedades hepáticas colestásicas.
CASO CLÍNICO
Paciente varón de 32 años, natural y procedente de Lima, casado, Testigo de Jehová, Chofer. Antecedentes de importancia: hepatomegalia desde recién nacido Fiebre Tifoidea a los 8 años. Alcohol: consumo social. Refiere que su hermana ha presentado, durante la gestación, 2 episodios de ictericia similares a los que el paciente presenta actualmente.
La historia de la enfermedad es de aproximadamente 10 años de curso episódico y recurrente con intervalos libres de enfermedad de duración variable. Los signos y síntomas iniciales fueron: ictericia y coluria, nauseas y vómitos, y disminución de peso no cuantificado. Es evaluado y tratado por un médico particular con fármacos no precisados y además tratamiento naturista luego del cual ceden los síntomas en aproximadamente 6 meses. Hace 5 años se agrega al cuadro clínico cansancio extremo durante el día e insomnio durante la noche además prurito intenso, el rascado produce excoriaciones de la piel que sangran fácilmente. Se ha repetido episodios de iguales síntomas mejorando siempre con tratamiento naturista.
Hace 3 semanas a la sintomatología descrita se agrega perdida ponderal cuantificada en 8 Kg. Niega nauseas y/o vómitos. Niega dolor abdominal. Por lo anterior acude a Consultorio externo de Gastroenterología del Hospital Nacional Arzobispo Loayza decidiéndose su hospitalización.
Niega haber consumido fármacos o algún tipo de hierba medicinal antes del inicio de la enfermedad.
Al Examen físico: regular estado general, mal estado nutricional, LOTEP, adelgazado, marcada ictericia de piel y escleras, huellas de rascado en piel, sin adenopatias periféricas ni edemas. El examen del tórax y cardiovascular no muestra alteraciones. Abdomen: RHA (+), blando, depresible, no doloroso a la palpación. Hígado palpable aproximadamente 4 cm debajo del RCD, de superficie lisa, consistencia conservada y borde romo.
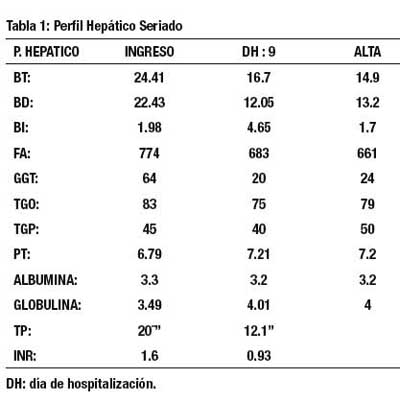
Exámenes auxiliares, destacan el hemograma que muestra Hb:10,3mg/dl. Valores normales de Glucosa, Urea y Creatinina. La serología para HIV y hepatitis virales (A,B y C) fueron Negativos. Beta 2 microglobulina y DHL normales. ANA, ANCAp, AMA, ASMA y Anti LKM – 1 todos negativos. El perfi l hepático mostró hiperbilirrubinemia a predominio directo, con fosfatasa alcalina elevada; llamando la atención la discreta elevación y la normalidad en el estudio seriado de la γ – glutamil transpeptidasa.(GGT) (Tabla 1).
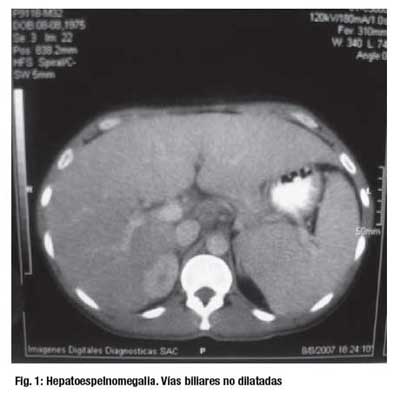
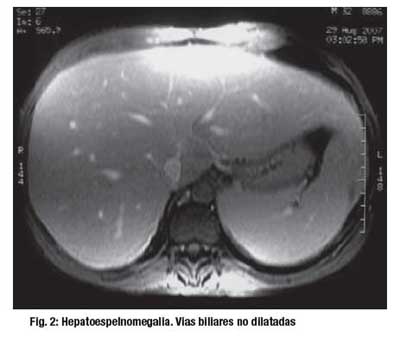
El estudio por Imágenes reveló: Ecografía abdominal, hepatomegalia con signos de hepatopatia difusa crónica e hipertensión portal, no evidenciándose dilatación de vías biliares intra o extrahepaitcas. TAC abdominal: aumento del volumen del lóbulo hepático izquierdo, esplenomegalia, Dilatación portal y de la vena esplénica relacionadas a hipertensión portal. Congestión de la vasculatura a nivel del mesenterio. (Figura 1). Colangioresonancia: hepatoesplenomegalia importante. Vías biliares intra y extrahepáticas normales. Páncreas sin alteraciones. Vesícula biliar con barro. (Figura 2).
EVOLUCIÓN
Se realizó biopsia hepática dirigida por laparoscopia, durante el procedimiento se observó el ligamento redondo hipervascularizado, el hígado aumentado de tamaño a expensas del lóbulo izquierdo que llega hasta el hipocondrio izquierdo, ambos lóbulos de superficie discretamente irregular y de coloración pardo-amarillenta. Se concluye: Hepatopatia crónica con esteatosis hepática, fibrosis incipiente y signos indirectos de hipertensión portal. Se toma biopsia del lóbulo izquierdo percibiéndose consistencia fibrosa leve.
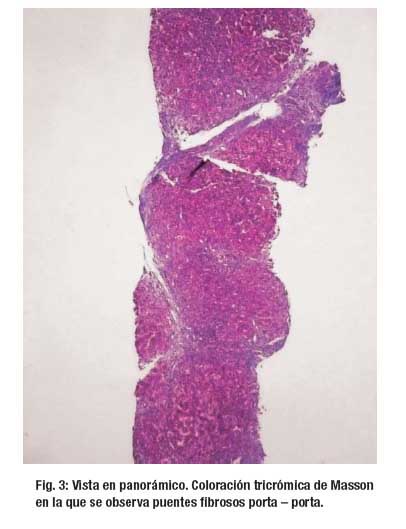
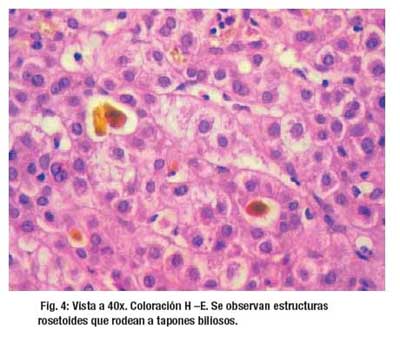
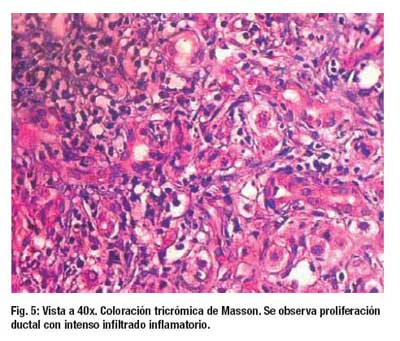
El examen microscópico de la biopsia muestra una arquitectura hepática distorsionada por la presencia de tractos fibrosis en puente porta – porta (2+). Se reconoce 11 espacios porta ampliados por fibrosis laxa, edema, células infl amatorias a predominio de linfocitos (1+), PMN (1+) y ocasionales eosinofilos que rebasan placa limitante; hay marcada proliferación ductal y metaplasia de la placa ductal que aun comprometen el area 1 acinar. Hepatocitos con degeneración moderada a severa y se disponen en estructuras rosetoides, hay colestasis, pigmento biliar intracitoplasmático, asi como abundantes tapones biliares a nivel canalicular, estos últimos vistos desde la zona 3 a 1 acinar. Hay también focos de necrosis con presencia de PMN, vena hepática terminal sin alteraciones.. (Figura 3, Figura 4 y Figura 5)
La endoscopia digestiva alta muestra várices esofágicas incipientes.
El paciente recibió acido ursodesoxicolico 250 mg VO tid desde el 2° día de hospitalización, además de un ciclo de tres días de vitamina K 10mg EV c/24h. Asimismo recibió una transfusión de 1 unidad de plasma fresco congelado el día 3 de hospitalización, previa a la realización de biopsia hepática por laparoscopia.
DISCUSIÓN
La Colestasis Intrahepatica Benigna Recurrente (CIBR) es una enfermedad caracterizada por episodios recurrentes y autolimitados de ictericia y prurito intensos, de duración variable de semanas a meses(6,7,8). Esta entidad fue reportada por primera vez hace más de 40 años en dos pacientes ingleses. Posteriormente se han reportado hasta el momento más de cien casos a nivel mundial provenientes de Europa, África, Norteamérica, Sudamérica y Japón(6). En nuestro país, también se ha reportado un caso de esta entidad(9).
A pesar de que la mayoría de casos de CIBR han sido descritos aisladamente, el descubrimiento de una historia familiar de colestasis hasta en el 50% de los pacientes, conduce tempranamente al estudio genético, demostrándose una transmisión autosómica recesiva de la enfermedad(6). Esta característica está presente en nuestro caso ya que en la historia familiar registrada tenemos una hermana del pacientes quien presentó 2 episodios de ictericia y prurito. La CIBR es frecuentemente subdiagnosticada en mujeres ya que en ellas la ictericia y el prurito síntomas característicos más comunes de esta enfermedad, son mal atribuidas a Colestasis inducida por el embarazo o por el uso de anticonceptivos orales(6).
Recientemente se ha logrado ubicar el defecto genético a nivel del brazo largo del cromosoma 18(6), dicho gen traduce una ATPasa (ATP8B1) que participa en la translocación de aminofosfolípidos a nivel del hepatocito, este paso es necesario para la secreción de sales biliares(7,8), por lo tanto este defecto produce la disminución de la secreción de sales biliares.
Con respecto a la presentación clínica, el primer ataque de ictericia y prurito ocurre típicamente durante la adolescencia o la tercera década de la vida(6,8). En nuestro caso el paciente refi ere haberlo presentado hacia los 22 años de edad. Cada ataque puede durar de semanas a meses con una media de duración de aproximadamente 3 meses(10). Los períodos libres de enfermedad, varían de meses a años(6).
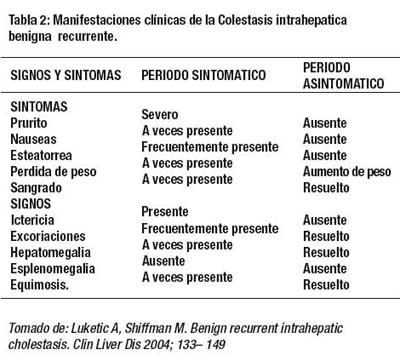
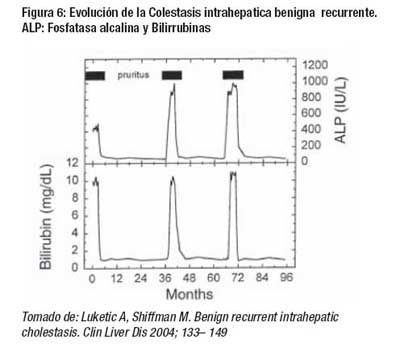
Las principales manifestaciones clínicas se presentan en la Tabla 2 y fig 6.
Los exámenes de laboratorio, durante los episodios sintomáticos revelan un patrón clásicamente colestásico con una elevación de la fosfatasa alcalina por lo menos doble del valor normal. La hiperbilirrubinemia es a predominio conjugado y puede elevarse a más de 10 veces el valor normal.
En contraste, las aminotransferasas estan normales o discretamente elevadas. La característica que distingue a esta entidad de otras formas de colestasis intrahepática familiar y de la diferencia de otras formas de colestasis intrahepática es el nivel de γ – glutamil transpetidasa, el cual permanece normal o ligeramente elevado(1,6,7,8,10). En nuestro paciente, este dato orientó al diagnóstico.
Los criterios diagnósticos de CIBR(6) se enumeran a continuación, siendo clara la concordancia de estos criterios con nuestro caso.
-
Como mínimo dos episodios de ictericia separados por un intervalo libre de síntomas que dure meses a años.
-
Valores de laboratorio consistentes con colestasis intra hepatica.
-
GGT normal o discretamente elevada.
-
Prurito severo secundario a colestasis.
-
Histología hepática demostrando colestasis centrolobular.
-
Vía biliar intrahepatica y extrahepatica normal, demostrado por colangiografia.
-
Ausencia de factores conocidos que se asocien a colestasis (drogas, embarazo, etc).
Como su nombre indica, clásicamente la evolución natural de la CIBR es benigna, sin progresión a fibrosis o insuficiencia hepática(6,7,8); característica que no se ajusta a nuestro caso en quien claramente se evidencia signos de insuficiencia hepática con hipertensión portal y fibrosis en la biopsia. Sin embargo, últimamente se han reportado casos de CIBR que progresan a Colestasis Intrahepatica Familiar Progresiva (CIFP)(8), ésta última caracterizada por evolución a fibrosis e insuficiencia hepática(7,8). Van Ooteghem y col., en un seguimiento de 63 pacientes con diagnóstico de CIBR reportaron que 4 de ellos desarrollaron un síndrome típico de Colestasis Intrahepatica Familiar Progresiva tipo 1 (CIFP1).
Estos pacientes progresaron a colestasis permanente hacia la segunda a cuarta década de la vida, y posterior insuficiencia hepática. Las biopsias hepáticas en estadios tardíos fueron más características de CIFP1, con fibrosis y formación de puentes porta – porta(11).
La Colestasis intrahepática familiar progresiva incluye a un grupo especifico de pacientes, clínicamente heterogéneo, requieren tener colestasis crónica recurrente para tener sospecha diagnostica, exclusión de enfermedades anatómicas o metabólicas identificables, patente de herencia autosómica recesiva y una combinación característica de aspectos clínicos bioquímicos e histológicos(1,7,8).
Esta colestasis familiar fue llamada hasta hace pocos años, enfermedad de Byler debido a que la primera descripción de la enfermedad se hizo en miembros descendientes de Jacob Byler perteneciente a una familia Amish. Clínicamente la enfermedad se caracteriza por ictericia, esteatorrea, retardo del crecimiento, y una actividad sérica disminuida o normal de gamaglutamiltranspeptidasa (GGT) a pesar de niveles aumentados de fosfatasa alcalina. La historia natural de la enfermedad es la progresión a la cirrosis y muerte por insuficiencia hepática(7,8,11)
Las bases fisiopatológicas de la CIFP están dadas por un defecto en la función excretora hepática, específicamente en la alteración del flujo biliar. El fenotipo de la CIFP expresa defectos o alteraciones en cualquiera de los genes que expresan proteínas importantes en la formación de la bilis(7,8).
En los últimos años estas proteínas transportadoras han sido identificadas, clonadas y caracterizadas funcionalmente lo que ha permitido caracterizar los síndromes de PFIC en tres tipos: (Tabla 3.)
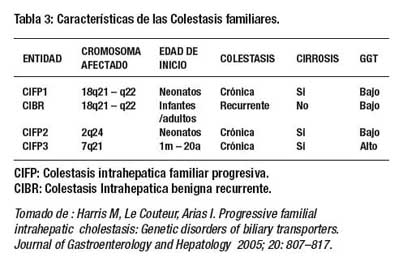
La CIBR ha sido mapeada en el mismo gen (18q21- 22) que la CIFP1. Se especula que puede haber un gen de colestasis familiar (FIC1) que es el responsable para ambas entidades a pesar de su diferente fenotipo y pronóstico( 7,8).
Por lo anterior, se puede plantear la hipótesis que ambas entidades son parte de una misma enfermedad, cuya progresión, a pesar de ser un raro evento, puede desarrollarse a cualquier edad, tanto temprana como tardíamente (11).
En nuestro caso, el cuadro clínico y la evolución natural de la enfermedad corresponden a Colestasis intrahepatica benigna recurrente; mientras que, la histología hepática concuerda con la Colestasis intrahepatica familiar progresiva tipo 1.
REFERENCIAS
1. DAVERN T, SCHARSCHMIDT B. Pruebas bioquímicas hepáticas. En: Feldman M, Friedman L, Sleisenger M. Enfermedades Gastrointestinales y Hepáticas. 7° ed. Buenos Aires: Editorial Médica Panamericana; 2004. p. 1300 – 1313.
2. MC GILL JM, KWIATKOWSKI AP. Cholestatic liver diseases in adults. Am J Gastroenterol 1998; 93: 684-91.
3. PÉREZ FERNÁNDEZ T, LÓPEZ SERRANO P, TOMÁS E, GUTIÉRREZ ML, LLEDÓ JL, CACHO G, SANTANDER C, FERNÁNDEZ RODRIGUEZ CM. Diagnostic and therapeutic approach to cholestatic liver disease. Rev Esp Enferm Dig 2004; 96: 60-73.
4. RESHEF R, SBEIT W, LACHTER L. The chronic cholestasis enigma in adults. IMAJ 2002; 4: 449-53.
5. BLUM H.Chronic cholestatic liver diseases. Journal of Gastroenterology and Hepatology (2002) 17, S399–S402.
6. LUKETIC A, SHIFFMAN M. Benign recurrent intrahepatic cholestasis. Clin Liver Dis 2004; 133– 149.
7. ELFERINK R. Cholestasis. Gut 2003;52 (Suppl II): II42–II48.
8. HARRIS M, LE COUTEUR, ARIAS I. Progressive familial intrahepatic cholestasis: Genetic disorders of biliary transporters. Journal of Gastroenterology and Hepatology 2005; 20: 807–817.
9. RAMOS R, FERNANDEZ M, ARAGON R. Colestasis intrahepatica recurrente benigna que evoluciona a cirrosis biliar secundaria. Resumen de las comunicaciones libres. XXVII Congreso Panamericano de Enfermedades Digestivas. Lima Perú. 2001.
10. BRENARD R, GEUBEL AP, BENHAMOU JP. Benign recurrent intrahepatic cholestasis. J Clin Gastroenterol 1989;11:546– 51.
11. VAN OOTEGHEM NA, KLOMP LW, VAN BERGEHENEGOUWEN GP, HOUWEN RH. Benign recurrent intrahepatic cholestasis progressing to progressive familiar intrahepatic cholestasis: low GGT cholestasis is a clinical continuum. J. Hepatol. 2002; 36: 439–43.
12. LEFKOWITCH J. Histological assessment of cholestasis. Clin Liver Dis 2004 8: 27– 40.
