










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Gastroenterología del Perú
Print version ISSN 1022-5129
Rev. gastroenterol. Perú vol.33 no.3 Lima July/Sep. 2013
REPORTE DE CASOS
Hepatopatía crónica asociada a fibrosis quística en el Hospital Alberto Sabogal Sologuren, Lima, Perú: reporte de caso
Chronic liver disease associated with cystic fibrosis in th Hospital Alberto Sabogal Sologuren, Lima, Peru: report of a case
Alexis Ormeño J. 1a, Cesar García D. 2b, Julia Sumire U. 3c, Carmen Asato H. 3c
1 Servicio de Gastroenterología Pediátrica, Instituto Nacional de Salud del Niño. Lima, Perú.
2 Servicio de Gastroenterología, Hospital Alberto Sabogal Sologuren. Lima, Perú.
3 Clínica Las Américas. Lima, Perú.
a Médico Residente, b Médico Asistente, c Médico Anátomo Patólogo
RESUMEN
La fibrosis quística (FQ) es la enfermedad genética recesiva más frecuente en la población caucásica y está producida por la alteración del transporte de electrolitos y agua en la membrana de las células epiteliales. La enfermedad hepática es una complicación frecuente hacia el final de la primera década de la vida, siendo raro su inicio posterior, excepto en pacientes con antecedente de íleo meconial. La lesión hepática característica en la FQ es la cirrosis biliar focal, aunque también se puede encontrar infiltración grasa. El diagnóstico se realiza considerando los hallazgos clínicos, laboratoriales y de imagen, teniendo en cuenta que las pruebas de función hepática normales no descartan la enfermedad. La ecografía es la técnica más ampliamente utilizada y permite evidenciar la presencia de esteatosis, litiasis, fibrosis, cirrosis, hipertensión portal o las alteraciones del árbol biliar. También están disponibles técnicas como la tomografía computarizada o la resonancia magnética, que permiten efectuar un estudio morfológico. Aspectos importantes en el tratamiento son el manejo nutricional, administración de vitaminas liposolubles y el uso de acido ursodesoxicólico (UDCA). En casos de cirrosis avanzada, el trasplante, aislado o combinado con el pulmonar, es una opción que cabe considerar, con tasas de supervivencia aceptables. Se presenta el caso de un paciente de 11 años de edad con diagnóstico de Hepatopatía crónica asociada a fibrosis quística.
Palabras clave:: Fibrosis quística; Hepatopatías; Cirrosis hepática biliar (fuente: DeCS BIREME).
ABSTRACT
Cystic fibrosis (CF) is the most frequent recessive genetic disorder in the caucasian population and is produced by the alteration of electrolyte and water transport in the epithelial cell membrane. Liver disease is a frequent complication towards the end of the first decade of life, being weird its onset, except in patients with a history of meconium ileus. The characteristic liver injury in CF is focal biliary cirrhosis, but fatty infiltration can also be found. The diagnosis is made considering the clinical, laboratory and imaging results having in consideration that the normal liver function tests do not rule out the disease. Ultrasound is the most widely used and can detect the presence of steatosis, stones, fibrosis, cirrhosis, portal hypertension or abnormalities of the biliary tree. There is an also available technique such as computed tomography or magnetic resonance imaging, which allows a morphological study. Important aspects in the treatment are nutritional management, administration of soluble vitamins and the use of ursodeoxycholic acid (UDCA). In cases of advanced cirrhosis, transplantation, isolated or combined with the lung, is an option to consider, with acceptable survival rates. We report the case of an 11 year old patient with a diagnosis of chronic liver disease associated with cystic fibrosis.
Key words: Cystic fibrosis; Liver diseases; Liver cirrhosis, biliary (source: MeSH NLM).
INTRODUCCIÓN
La fibrosis quística (FQ) es la enfermedad genética recesiva más frecuente en la población caucásica. Está producida por la alteración del transporte de electrolitos y agua en la membrana de las células epiteliales (1)).
De naturaleza hereditaria, el defecto se debe a mutaciones del gen 7q31.2, que codifica para una proteína (regulador transmembrana de fibrosis quística [CFTR]) (2) expresada en la membrana de las células epiteliales de las glándulas mucosas del tracto respiratorio, digestivo y reproductor y en las glándulas serosas del sudor y la saliva; y que se comporta como un canal de cloro regulado por AMP-c. La mutación más frecuente se debe a la pérdida del aminoácido fenilalanina en el codón 508(F508del) (3). El hecho patogénico fundamental es la afectación de las funciones de las glándulas exocrinas y un transporte transepitelial anómalo de iones, que dan lugar a secreciones anormalmente espesas y viscosas, lo que favorece la obstrucción y, más tarde, facilita la colonización bacteriana que altera el funcionamiento normal de múltiples órganos y sistemas (4)).
La FQ es una enfermedad multiorgánica, caracterizada clásicamente por una tríada clínica que incluye: afectación respiratoria (enfermedad pulmonar obstructiva crónica con bronquiectasias), insuficiencia pancreática y la prueba del sudor alterada. Sin embargo, afecta potencialmente a todos los tejidos epiteliales, incluyendo manifestaciones clínicas tan diversas como pólipos nasales, azoospermia, alteraciones biliares y digestivas (1).
La enfermedad hepática es relativamente habitual y aparece como una complicación temprana. La lesión típica es la cirrosis biliar focal (CBF), causada por una obstrucción biliar y una fibrosis periportal progresiva, pero en la mayoría de los casos se desarrollan alteraciones hepáticas leves con una discreta elevación de las pruebas bioquímicas y áreas focales de afectación del tracto portal con obstrucción biliar, proliferación de los conductillos biliares y colangitis y sólo una pequeña proporción de pacientes desarrollan una alteración severa con cirrosis multilobular (CBM), que conduce a la aparición de hipertensión portal (HP) e hiperesplenismo (5,6). La etiopatogenia es multifactorial, con determinantes genéticos y ambientales.
Se presenta el caso de un paciente con diagnóstico de Hepatopatía Crónica por Fibrosis Quística.
CASO CLÍNICO
Paciente varón de 11 años de edad natural y procedente de Lima con diagnóstico de fibrosis quística desde los 9 años de edad. En la tomografía espiral multicorte torácica de control (enero 2012), incidentalmente se observa un patrón nodular del parénquima hepático. Por este motivo se decide su hospitalización para realizar una biopsia hepática ante la sospecha de hepatopatía por fibrosis quística.
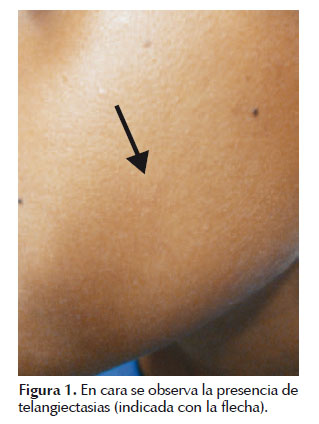
Al examen físico se observó la presencia de telangiectasias en cara (Figura 1) e hipocratismo digital. En el aparato respiratorio a la auscultación se encontró roncantes y sibilantes espiratorios en ambos hemotórax. No se palparon visceromegalias.
Como antecedente,el paciente es producto de quinta gestación, parto vaginal, a término, macrosómico (4,100 gr). No se reportó presencia de íleo meconial. A los 3 meses de edad fue hospitalizado por un mes con el diagnóstico de neumonía y síndrome obstructivo bronquial agudo recibiendo durante su hospitalización 15 días de antibioticoterapia, corticoide y salbutamol inhalados. La madre refiere un desarrollo psicomotor y ganancia ponderal adecuadas.
Desde el año de edad presentó deposiciones diarias, duras y en ocasiones caprinas. A los 3 años de edad presentó un episodio de diarrea persistente de 1 mes de duración con esteatorrea en número de 4 a 5 deposiciones diarias sin fiebre, que remitió con tratamiento antibiótico sin ameritar hospitalización. Desde los 4 años presenta episodios de tos matutina diariamente que remitía parcialmente con el uso de salbutamol inhalado. A los 7 años la tos fue en incremento acompañado de dificultad respiratoria siendo necesario nebulizaciones con fenoterol así como uso mas frecuente de salbutamol. Alos 8 años es evaluado por mayor necesidad de ingresos por emergencia para nebulización y al examen físico evidencian hipocratismo digital y en los estudios por imágenes (TEM tórax) se encontró la presencia de bronquiectasias y se plantea la posibilidad de fibrosis quística, siendo corroborado con test de sudor positivo. Desde los 9 años recibe corticoides inhalados (Fluticasona) con disminución de episodios de exacerbación de tos y necesidad de nebulizaciones.
Antecedentes familiares no contributorios.
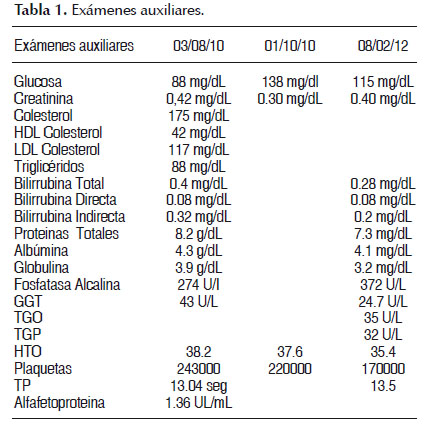
Los exámenes auxiliares se muestran en la Tabla 1.
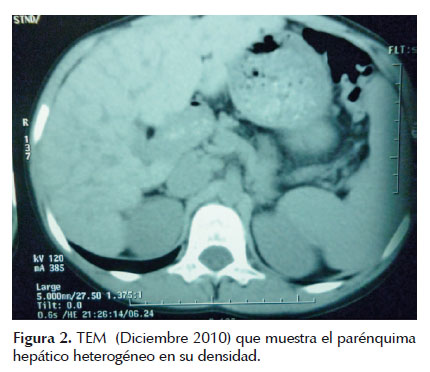
En diciembre del 2010 se realizó una TEM de tórax donde incidentalmente se observó heterogeneidad en la densidad del parénquima hepático (Figura 2).
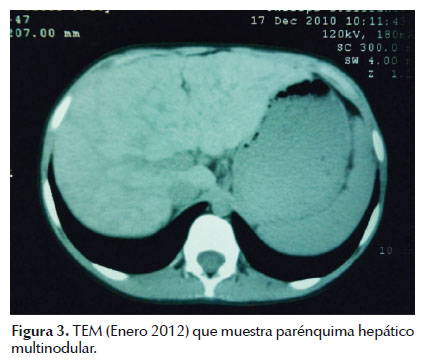
En enero del 2012 la TEM torácica de control mostró el parénquima hepático de aspecto multinodular (Figura 3).
La biopsia hepática confirmó la presencia de hepatopatía con arquitectura hepática parcialmente distorsionada por la presencia de puentes fibrosos y nódulos de regeneración incompletos rodeados de bandas fibrosas (Figura 4).
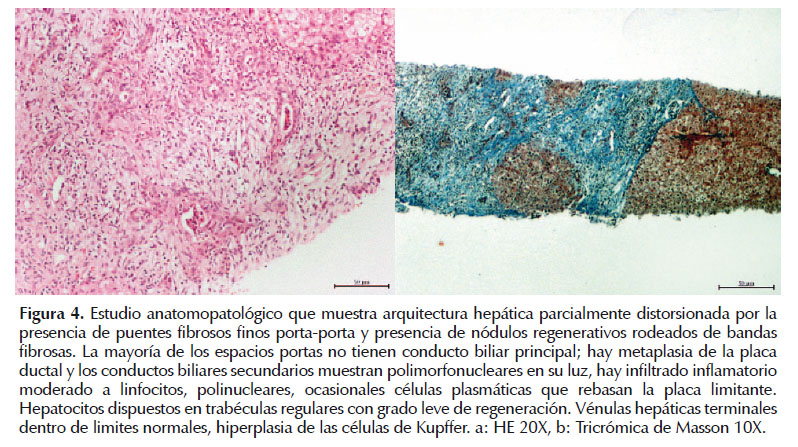
DISCUSIÓN
A pesar que la enfermedad hepática crónica como complicación de la FQ ya fue descrita en 1938 por Andersen (7), la importancia de esta patología ha sido relegada por otros signos y síntomas más obvios a nivel respiratorio y pancreático, a pesar de ser actualmente la segunda causa de muerte en los pacientes con FQ. No se conoce bien la incidencia y la prevalencia de la enfermedad hepática, al no disponerse de marcadores diagnósticos sensibles y específicos, y se considera que si hay un diagnóstico de sospecha, tras la valoración clínica y bioquímica y mediante técnicas de imagen, las cifras se sitúan entre el 18 y el 37%. En una revisión realizada en varios hospitales de España se constató hace unos años una frecuencia del 20,1%, y la edad media de presentación de la enfermedad fue de 7,2 años. La CBM aparece en un 5-15% de los casos y la prevalencia de HP es del 8% (8-10). Así pues, se puede afirmar que, si bien la enfermedad hepática es una complicación relativamente frecuente, la significación pronóstica de la HP supone sólo un 2,5% de la mortalidad total por FQ.
Los estudios de prevalencia sugieren que la enfermedad hepática asociada a FQ, clínicamente evidente, se desarrolla antes o durante la pubertad (11) mientras que la incidencia disminuye rápidamente después de los 10 años de edad (12) y aparece raramente después de los 18 años (13,14). Entre otros factores de riesgo para desarrollar afectación hepática se consideran la insuficiencia pancreática exocrina (IPE), el antecedente de íleo meconial, el sexo masculino y las mutaciones severas (10).
No se ha reportado relación entre el fenotipo de FQ y el desarrollo o la severidad de la enfermedad hepática, y el hecho de que no todos los pacientes presenten hepatopatía y que tanto su comienzo como su severidad sean variables sugiere que otros genes, denominados modificadores y determinados factores ambientales, pueden influir en su desarrollo. Algunos estudios recientes muestran que la asociación de determinados polimorfismos en los genes que regulan la inflamación, la fibrosis o el estrés oxidativo, como la alfa-1-antiproteasa ( -1-AT), el factor transformador del crecimiento (TGFB1), la glutatión-S-transferasa (GSTP1) y la lectina transportadora de manosa (MBL2), de modo aislado o asociados, aumenta significativamente el riesgo de desarrollar dicha afección y se ha asociado a una enfermedad grave (15). En un estudio multicéntrico (16) realizado en Estados Unidos y Canadá, se ha concluido que el gen alfa-antiproteasa (alelo Z) supone un riesgo 5 veces mayor de desarrollar una enfermedad hepática con HP y en otro estudio (17) realizado en Italia se señala el haplotipo MBL2 y el ABCB4 como moduladores del riesgo de enfermedad.
La lesión hepática característica en la FQ es la cirrosis biliar focal, aunque también se puede encontrar infiltración grasa. En el hígado, el CFTR se localiza en la membrana apical de los conductos biliares intrahepáticos: la secreción de cloruro defectuoso inhibe la hidratación de la bilis producida en el canalículo llevando a la obstrucción de la bilis intrahepatica. Además, las células epiteliales biliares intrahepáticas producen excesivo moco compuesto de proteoglicanos que contribuye aún más a la viscosidad de la bilis. Esta situación expone a los hepatocitos a una alta concentración de ácidos biliares potencialmente tóxicos que conducen a inflamación y cicatrización (18).
El diagnóstico de esta enfermedad (5,10,19) se realiza considerando los hallazgos clínicos (hepatomegalia o signos de disfunción hepática), laboratoriales (alteración de las enzimas y otras pruebas de función hepática) y de imagen. Las pruebas de función hepática normales no descartan la enfermedad, como en el caso de nuestro paciente. La ecografía (20) es la técnica más ampliamente utilizada y permite poner en evidencia la presencia de esteatosis, litiasis, fibrosis, cirrosis, hipertensión portal o las alteraciones del árbol biliar. Existen puntuaciones para valorar el grado de afectación; así, Williams et al. (21) basan la suya en la estructura (parénquima), la ecogenicidad (fibrosis periportal) y el borde hepático. También están disponibles técnicas como la tomografía computarizada o la resonancia magnética, que permiten efectuar un estudio morfológico (22,23). La gammagrafía es de utilidad para evaluar la función hepática y la excreción biliar y la colangio resonancia parece detectar con mayor precocidad la afectación biliar, tanto extrahepática como intrahepática. La endoscopía digestiva alta es una prueba muy sensible para detectar las complicaciones de la hipertensión portal como várices, gastropatía, etc.
La utilidad de la biopsia hepática es controvertida, ya que a pesar de detectar alteraciones tempranas e informar sobre el tipo de lesión, tiene el inconveniente de ser una técnica invasiva que, además, puede proporcionar unos resultados no representativos debido a un error de obtención de la muestra, al ser una lesión parcheada. No obstante, aporta una información importante y está indicada en casos dudosos o para establecer el diagnóstico de cirrosis.
El hígado puede ser morfológicamente normal. Histológicamente, se han descrito tres tipos de enfermedad hepática en pacientes con fibrosis quística: esteatosis, fibrosis y la cirrosis biliar focal multinodular. La infiltración grasa hepática es el hallazgo más común en los estudios de imágenes del hígado. También se observó en el 30% de las biopsias y hasta en 60% de autopsias de pacientes con FQ (16,17). Un hígado hiperecogénico en forma parcial o total es sugestivo de esteatosis en la ecografía.
Más recientemente, se han estudiado otros métodos de diagnóstico. Se ha trabajado en los componentes séricos del remodelado de la matriz hepática en el laboratorio y se pueden determinar los marcadores precoces de fibrosis (hay que tener en cuenta que la lesión más importante es la obstrucción biliar y la fibrosis): alfa-glutatión-S-transferasa A1 (GSTA), ácido hialurónico, colágena VI, IGFPB3, inhibidor de la metaloproteinasa de la matriz (TIMP-1), colágena IV, prolil-hidroxilasas y metaloproteinasa MMP-2. Estos marcadores tienen un valor potencial como determinantes precoces de lesión hepática. Otra herramienta diagnóstica es el FibroScan (24)), basado en la combinación de la ecografía y el estudio de la rigidez-elasticidad del hígado mediante la emisión de ondas de choque mecánicas que, en función de como se propaguen en el tejido, identifican los diferentes grados de elasticidad hepática y, por tanto, de fibrosis.
En cuanto al tratamiento de la FQ la optimización de la nutrición siempre es importante, pero en presencia de enfermedad hepática es una prioridad. La malnutrición es un factor de riesgo para el desarrollo de la enfermedad hepática, pero ésta por si misma contribuye a la anorexia y un mayor deterioro del estado nutricional. Se recomienda un aporte energético de 120 a150% por encima de las necesidades promedio estimadas para la edad y somatometría. Esto con el fin de superar los efectos del aumento del catabolismo asociado con la patología respiratoria y hepática asociada a la FQ.
Se deben administrar vitaminas liposolubles para maximizar los efectos antioxidantes y para evitar efectos secundarios tales como osteodistrofia ósea y hemorragia dependiente de déficit de vitamina K.
Los ácidos biliares, por lo general en forma de ácido ursodesoxicólico (UDCA), constituye la única opción farmacéutica para tratar una enfermedad hepática por FQ. UDCA es un antiinflamatorio que también estimula la secreción de iones de cloruro y bicarbonato colangiocelulares dependientes del calcio, favoreciendo así el flujo de bilis.
En pacientes con cirrosis muy avanzada, el trasplante, aislado o combinado con el pulmonar, es una opción que cabe considerar, con tasas de supervivencia aceptables (del 85% al año y del 64% a los 5 años) y efectos beneficiosos sobre la función pulmonar, el estado nutricional, la composición corporal y la calidad de vida en muchos casos, aunque algunos autores presentan cifras más bajas, e incluso se discute sobre la idoneidad de su indicación y el momento de hacerlo (10,25). Otros autores defienden la realización del procedimiento precozmente (cuando exista HP, pero antes de presentar descompensaciones severas), y algunos prefieren esperar a que exista una disfunción hepatocelular.
BIBLIOGRAFÍA
1. Tabernero da Veiga S, González Lama Y, Lama More R, Martínez Carrasco MC, Antelo Landeria MC, Jara Vega P. Hepatopatía crónica asociada a fibrosis quística: gasto
energético en reposo, factores de riesgo y repercusión en la evolución de la enfermedad. Nutr Hosp. 2004;19(1):19-27. [ Links ]
2. Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245(4922):1073-80. [ Links ]
3. Gradient of distribution in Europe of the major CF mutation and of its associated haplotype. European Working Group on CF Genetics (EWGCFG). Hum Genet. 1990;85(4):436-45. [ Links ]
4. Luder E. Cystic fibrosis the influence of the genotype and phenotype relationship on nutritional status. Top Clin Nutr. 2003;18(2):92-9. [ Links ]
5. Sokol RJ, Durie PR. Recommendations for management of liver and biliary tract disease in cystic fibrosis. Cystic Fibrosis Foundation Hepatobiliary Disease Consensus Group. J Pediatr Gastroenterol Nutr. 1999;28 Suppl 1:S1-13. [ Links ].
6. Shapira R, Hadzic N, Francavilla R, Koukulis G, Price JF, Mieli-Vergani G. Retrospective review of cystic fibrosis presenting as infantile liver disease. Arch Dis Child. 1999;81(2):125-8. [ Links ]
7. Andersen D. Cystic fibrosis of the pancreas and its relation to celiac disease a clinical and pathologic study. Am J Dis Child. 1938;56(2):344-99. [ Links ]
8. Corbett K, Kelleher S, Rowland M, Daly L, Drumm B, Canny G, et al. Cystic fibrosis-associated liver disease: a population-based study. J Pediatr. 2004;145(3):327-32. [ Links ]
9. Colombo C, Russo MC, Zazzeron L, Romano G. Liver disease in cystic fibrosis. J Pediatr Gastroenterol Nutr. 2006;43 Suppl 1:S49-55. [ Links ]
10. Colombo C. Liver disease in cystic fibrosis. Curr Opin Pulm Med. 2007;13(6):529-36. [ Links ]
11. Diwakar V, Pearson L, Beath S. Liver disease in children with cystic fibrosis. Paediatr Respir Rev. 2001;2(4):340-9. [ Links ]
12. Colombo C, Battezzati PM, Crosignani A, Morabito A, Costantini D, Padoan R, et al. Liver disease in cystic fibrosis: a prospective study on incidence, risk factors, and outcome.
Hepatology. 2002;36(6):1374-82. [ Links ]
13. Nash KL, Allison ME, McKeon D, Lomas DJ, Haworth CS, Bilton D, et al. A single centre experience of liver disease in adults with cystic fibrosis 1995-2006. J Cyst Fibros. 2008;7(3):252-7. [ Links ]
14. Bhardwaj S, Canlas K, Kahi C, Temkit M, Molleston J, Ober M, et al. Hepatobiliary abnormalities and disease in cystic fibrosis: epidemiology and outcomes through adulthood. J
Clin Gastroenterol. 2009;43(9):858-64. [ Links ]
15. Salvatore F, Scudiero O, Castaldo G. Genotype-phenotype correlation in cystic fibrosis: the role of modifier genes. Am J Med Genet. 2002;111(1):88-95. [ Links ]
16. Bartlett JR, Friedman KJ, Ling SC, Pace RG, Bell SC, Bourke B, et al. Genetic modifiers of liver disease in cystic fibrosis. JAMA. 2009;302(10):1076-83. [ Links ]
17. Tomaiuolo R, Degiorgio D, Coviello DA, Baccarelli A, Elce A, Raia V, et al. An MBL2 haplotype and ABCB4 variants modulate the risk of liver disease in cystic fibrosis patients: a
multicentre study. Dig Liver Dis. 2009;41(11):817-22. [ Links ]
18. Taylor CJ, Connolly S. Hepatobiliary disease in cystic fibrosis. Paediatrics an Child Health. 2009;20(1):20-5. [ Links ]
19. Moyer K, Balistreri W. Hepatobiliary disease in patients with cystic fibrosis. Curr Opin Gastroenterol. 2009;25(3):272-8. [ Links ]
20. Mueller-Abt PR, Frawley KJ, Greer RM, Lewindon PJ. Comparison of ultrasound and biopsy findings in children with cystic fibrosis related liver disease. J Cyst Fibros. 2008;7(3):215-21. [ Links ]
21. Williams SG, Evanson JE, Barrett N, Hodson ME, Boultbee JE, Westaby D. An ultrasound scoring system for the diagnosis of liver disease in cystic fibrosis. J Hepatol. 1995;22(5):513-21. [ Links ]
22. King LJ, Scurr ED, Murugan N, Williams SG, Westaby D, Healy JC. Hepatobiliary and pancreatic manifestations of cystic fibrosis: MR imaging appearances. Radiographics.
2000;20(3):767-77. [ Links ]
23. Akata D, Akhan O, Ozcelik U, Ozmen MN, Oguzkurt L, Haliloglu M, et al. Hepatobiliary manifestations of cystic fibrosis in children: correlation of CT and US findings. Eur J
Radiol. 2002;41(1):26-33. [ Links ]
24. Witters P, De Boeck K, Dupont L, Proesmans M, Vermeulen F, Servaes R, et al. Non-invasive liver elastography (Fibroscan) for detection of cystic fibrosis-associated liver disease. J Cyst Fibros. 2009;8(6):392-9. [ Links ]
25. Nash KL, Collier JD, French J, McKeon D, Gimson AE, Jamieson NV, et al. Cystic fibrosis liver disease: to transplant or not to transplant? Am J Transplant. 2008;8(1):162-9. [ Links ]
Correspondencia:
Dr. Alexis Ormeño J.
E-mail: alexisojulca@gmail.com
Recibido: 01/03/2013
Aprobado: 23/08/2013
