










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Gastroenterología del Perú
Print version ISSN 1022-5129
Rev. gastroenterol. Perú vol.34 no.4 Lima Oct. 2014
Caracterización molecular de cáncer colorrectal hereditario en Perú
Molecular characterization of hereditary colorectal cancer in Peru
Cesar Ñique Carbajal 1, Fernando Sánchez Renteria 2, Barbara Lettiero 3, Patrik Wernhoff 4, Mev Domínguez-Valentin 5
1 Departamento de Ciencias de la Salud, Universidad Católica Santo Toribio de Mogrovejo. Lambayeque, Perú.
2 Estudiante Escuela de Medicina, Universidad Católica Santo Toribio de Mogrovejo. Lambayeque, Perú.
3 Department of Oncology-Pathology, Institute of Clinical Sciences, Lund University. Lund, Sweden.
4 Department of Experimental Medical Science, Unit of Muscle Biology, Lund Transgenic Core Facility/Reproductive Immunology, Lund University. Lund, Sweden.
5 Department of Biomedicine, University of Bergen, 5009 Bergen, Norway.
RESUMEN
Objetivo: Investigar molecularmente la deficiencia en los genes de reparo de DNA (MMR) asociados al síndrome de Lynch. Materiales y métodos: Realizar los análisis de inmunohistoquímica e inestabilidad de microsatélites (MSI) en 5 familias con sospecha de síndrome de Lynch de acuerdo a los criterios clínicos de Amsterdam y/o Bethesda, atendidos en el Hospital Nacional Almanzor Aguinaga Asenjo de la ciudad de Chiclayo (Lambayeque-Perú) durante 2007-2010. Resultados. La falta de expresión de las proteínas MLH1/PMS2 y una alta MSI (MSI-H) fueron observados en un paciente de sexo masculino de 60 años diagnosticado con adenocarcinoma de grado I. Adicionalmente, se realizó el análisis mutacional puntual en el gen BRAF (V600E) a fin de descartar que se trate de un caso esporádico de cáncer colorrectal. La ausencia de mutación en el gen analizado asociado a los resultados moleculares del tumor, sugiere la caracterización de este paciente como sospecha de síndrome de Lynch. Conclusiones: Es el primer estudio molecular reportado en la población peruana y demuestra la importancia del análisis molecular en familias con sospecha de cáncer colorrectal hereditario a fin de ofrecer posibilidades de vigilancia y seguimiento que han demostrado reducir la morbilidad y la mortalidad del cáncer colorrectal así como contribuir a la caracterización a nivel genética y clínica de este tipo de cáncer en la población peruana.
Palabras clave: Cáncer colorrectal; Síndrome de Lynch; Genética; Perú (fuente: DeCS BIREME).
ABSTRACT
Objective. To investigate the molecular deficiency in MMR genes associated to Lynch syndrome. Material and methods: Immunohistochemical and microsatellite instability (MSI) analysis were performed in 5 families with suspected Lynch syndrome according to the clinical criteria, Amsterdam and/or Bethesda that had been treated at the Hospital Nacional Almanzor Aguinaga Asenjo (Lambayeque-Peru) during 2007-2010. Results: The absence of expression of MLH1/PMS2 and high MSI (MSI-H) were observed in a male patient aged 60 with adenocarcinoma grade I. In addition, the point mutational analysis was performed in BRAF (V600E) to rule that it is a sporadic case of colorectal cancer. The absence of mutation in BRAF together with the molecular analysis suggests the suspicion as a Lynch syndrome. Conclusions: It is the first molecular study reported in the Peruvian population and demonstrates the importance of molecular analysis in families with suspected hereditary colorectal cancer in order to provide possibilities of surveillance and monitoring that have been shown to reduce morbidity and mortality of colorectal cancer. The present study contributes to the genetic and clinical characterization of the Lynch syndrome in the Peruvian population.
Key words: Colorectal neoplasms; Lynch syndrome; Genetic; Peru (source: MeSH NLM).
INTRODUCCIÓN
El síndrome de Lynch (SL) representa aproximadamente 4% de todos los tipos de cáncer colorrectal (1). El SL es una enfermedad hereditaria con patrón autosómico dominante que se caracteriza por un riesgo aumentado del desarrollo de cáncer colorrectal (CCR) a edades más tempranas en comparación con los casos de CCR esporádicos (2). El síndrome de Lynch es causado por mutaciones germinales en los genes reparadores del ADN (MMR): MLH1 (42%), MSH2 (33%), MSH6 (18%) y PMS2 (8%) (3). Con el fin de estratificar a las familias para el análisis genético, se definieron los criterios de Amsterdam (CA) basado en el desarrollo de cáncer colorrectal (CA-I) o en la coocurrencia de tumores extracolónicos en las familias con síndrome de Lynch (CA-II). Posteriormente se incluyó la evaluación molecular para identificar a los pacientes con sospecha de SL (Directrices revisadas de Bethesda) (4-6).
El rastreamiento molecular de pacientes con sospecha de SL permite revelar de forma indirecta la presencia de mutaciones en la línea germinal y comprende la inestabilidad de microsatélites (MSI) y la inmunohistoquímica (IHQ), estudiados a partir del tejido tumoral colorectal (7-11). La secuenciación de los genes de MMR es la técnica definitiva y confirmatoria de los resultados proporcionados por la IHQ y la MSI (12).
El estudio mutacional de los genes de MMR en familias con sospecha de síndrome de Lynch de América del Sur identificó 37% (rango 27-65% en diferentes países/registros) de las mutaciones en los genes MLH1 y MSH2 en familias que cumplen con los criterios de Amsterdam y/o Bethesda (9,13). Los registros de cáncer colorrectal hereditario han sido recientemente establecidos en Argentina, Brazil, Uruguay y Chile con la finalidad de recolectar, compartir información sobre el espectro mutacional de los genes de MMR, identificar mutaciones fundadoras potenciales, interpretar el rol de las variantes genéticas de significancia desconocida (VUS) así como analizar los riesgos del desarrollo de cáncer hereditario en la población de América del Sur, sin embargo el diagnostico genético de SL aún no está disponible en el sistema público de salud (13).
El objetivo del presente estudio es investigar molecularmente pacientes con sospecha clínica de SL, caracterizar molecular y genéticamente esta población a fin de definir la mejor estrategia de estudio aplicable a este grupo de pacientes en un país con recursos limitados como Perú.
MATERIALES Y MÉTODOS
Consentimiento ético
Todos los pacientes otorgaron su consentimiento informado para la inclusión voluntaria en el estudio. El Comité de Bioética de la Facultad de Medicina de la Universidad Católica Santo Toribio de Mogrovejo (USAT) aprobó la investigación según Resolución N° 016 -2011 CB FM USAT.
Selección de pacientes
Los pacientes fueron seleccionados de acuerdo al cumplimiento de los criterios clínicos de Amsterdam y/o Bethesda. La información clínica y patológica fueron recopiladas según el reporte del Boletín de Cancerología de la Oficina de Epidemiología del Hospital Almanzor Aguinaga Asenjo entre los años 2007 – 2010, la historia familiar de los pacientes que se sospecha tendrian SL fue recolectada a través de una entrevista personal.
Extracción de ADN
La extracción del ADN genómico para el análisis mutacional de la línea germinal fue realizado a partir de una muestra de sangre periférica (3 a 5 gotas de sangre del pulpejo de los dedos) colectada en papel filtro Whatman N° 3 siguiendo el protocolo de extracción de QiampDNA Mini Kit (Qiagen Valencia, CA).
Las áreas no necróticas tumorales del tejido tumoral fijado en formalina y embebido en parafina (FFPE) fueron macrodisectadas. La extracción del ADN tumoral fue realizada a partir de tres secciones de 5-mm de la muestra de FFPE usando el Kit FFPE Qiagen (Qiagen Valencia, CA) de acuerdo a las instrucciones de los fabricantes. La concentración de ADN fue determinado mediante el Qubit Fluorometric Quantitation (Invitrogen) y las muestras con suficiente y alta calidad de ADN fueron seleccionadas para los analisis de MSI.
MSI e IHQ
El análisis de MSI fue realizado mediante el Sistema de Análisis MSI, Versión 1.2 (Promega, Madison, WI). El ADN extraído y amplificado fue analizado en el Secuenciador Automático 3130XL (Applied Biosystems, Foster City, CA). El análisis incluyó 5 marcadores mononucleotidos BAT-25 BAT-26, NR-21, NR-24 y MONO-27 (Promega, MSI Analysis System, Version 1.2, Madison, WI). Los resultados fueron evaluados mediante el GeneMapper Software Version 4.0 (Applied Biosystems, Foster City, CA) y definido como alta-MSI (MSI-H) cuando ≥2 marcadores son inestables, baja-MSI (MSI-L) cuando 1 marcador es inestable y estable (MSS) cuando ninguno de los marcadores mostró inestabilidad.
La IHQ se realizó para las 4 proteínas de MMR, MLH1, PMS2, MSH2 y MSH6 a partir de los tumores colectados. Brevemente, 4-µm secciones fueron colocados en los slides microscopicos SuperFrost® Plus. La recuperación de los antigenos fue realizado en un hervidor a presión en Solución de Recuperación Antigeno Especifica, pH 9 (Dako, Glostrup, Denmark) y teñidos en un immunostainer automatico (Autostainer Plus, Dako, Glostrup, Denmark) usando el Dako EnVisionFLEX+ Detection System, Peroxidase/DAB, Rabbit/Mouse (Dako, Glostrup, Denmark) de acuerdo a las instrucciones de los fabricantes. Los anticuerpos utilizados fueron MLH1, clone ES05 (Dako, Glostrup, Denmark, dilución 1:100), PMS2, clone A16-4 (BD Pharmingen, San Diego, CA, dilución 1:300), MSH2, clone FE11 (Calbiochem, Merck KgaA, Darmstadt, Germany, dilución 1:100), y MSH6, clone EPR3945 (Epitomics, Burlingame, dilución 1:100). La expresión tumoral de MMR fue establecida como normal (retenida), pérdida y débil (cuando la intensidad de la IHQ es reducida al ser comparada con el control interno normal).
Secuenciación
El análisis de la mutacion puntual en el gen BRAF (V600E) fue realizado mediante la amplificación por PCR y secuenciada utilizando Illumina Genome Analyzer (Illumina) de acuerdo a las instrucciones del fabricante. La secuenciación de los genes de MMR fue no realizada debido a la insuficiencia de ADN genómico.
RESULTADOS
Cinco pacientes fueron seleccionados de acuerdo a los criterios clínicos de Amsterdam y/o Bethesda, 3/5 tuvieron material suficiente para realizar los análisis de MSI e IHC pero insuficiente para la secuenciación de los genes de MMR. Las características clínicas y resultados moleculares de los pacientes están descritas en la Tabla 1.

La extracción del ADN, MSI, IHQ y la secuenciación fueron realizadas gracias al apoyo del Departamento de Oncología del Instituto de Ciencias Clínicas de la Universidad de Lund (Lund, Suecia). 1/3 (33%) de los pacientes analizados presentó ausencia de expresión de las proteínas MLH1/PMS2 y una MSI-H (Figura 1 y 2). Estos marcadores moleculares alterados sugirieron el análisis de la mutación puntual en el gen BRAF (V600E) que es un marcador de casos esporádicos. El resultado negativo para la mutación puntual confirma que no se trata de un tumor colorrectal esporádico, sugiriendo como un caso de sospecha de SL.
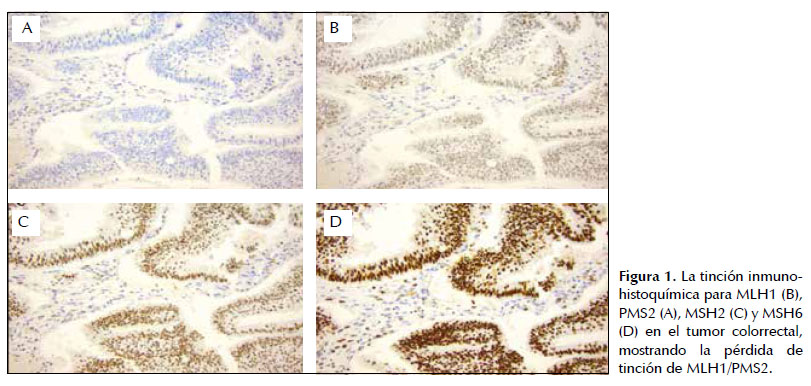
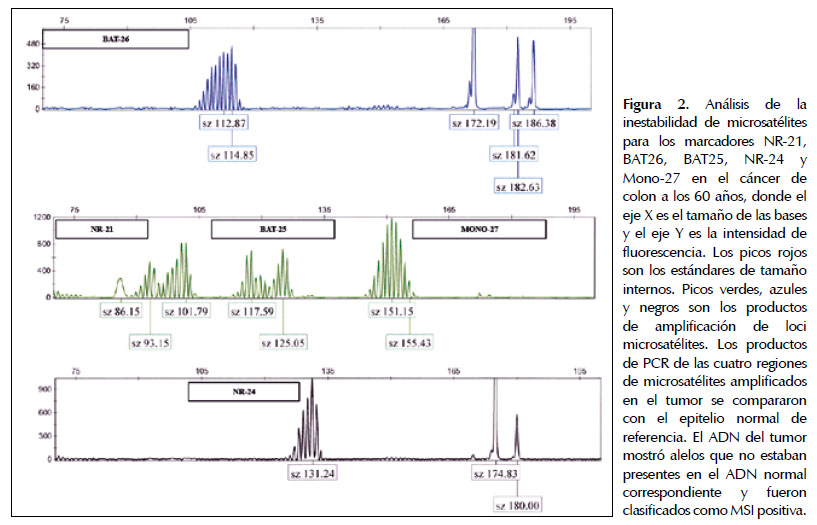
El paciente en mención es de sexo masculino, residente en la ciudad de Chiclayo, con antecedentes personales de epilepsia por neurocisticercosis, estreñimiento crónico, tumor maligno de colon diagnosticado a la edad de 60 años, ausencia de diabetes mellitus e hipertensión arterial. Los antecedentes familiares que presenta el paciente comprenden un hermano de primer grado con cáncer de colon diagnosticado a la edad de 55, con 3 hijas aparentemente sanas al momento de la entrevista. El diagnostico anatomopatologico determinó el adenocarcinoma con componente mucinoso menor de 50% bien diferenciado con invasión a la muscular propia (PT2).
DISCUSIÓN
El SL es un síndrome de predisposición al cáncer hereditario de colon más común. Es un síndrome autosómico dominante causado por mutaciones en la línea germinal en los genes MLH1, MSH2, MSH6 y PMS2. El SL representa el 2-3% de todos los casos diagnosticados de cáncer colorrectal y el 5-9% de cáncer de endometrio en pacientes menores de 50 años de edad (1-2). En nuestro estudio, el número de pacientes con sospecha de Síndrome de Lynch es de 33% (1/3), este porcentaje se encuentra en el rango (27-65%) de las familias sudamericanas diagnosticadas molecular y clínicamente con síndrome de Lynch (9,1315). Sin embargo, esta tasa es alta al ser comparado con las tasas de prevalencia de mutación identificada para MLH1 (28%) y para MSH2 (18%) en la poblacion de Asia; 31% y 20% en la población multi-etnica americana; 26% y 19% en las poblaciones Europeas y Australianas (16).
El presente estudio confirma la sensibilidad de IHQ e MSI para la detección de casos con sospecha de Síndrome de Lynch, en concordancia con Boland et al., quien considera la MSI como un marcador molecular para la identificación de familias con SL, asi como marcador de pronostico y predicción (17). Recientemente, Van Lier et al. mediante un estudio poblacional multicéntrico prospectivo determinó el uso de la MSI para todos los pacientes diagnosticados con cáncer colorrectal hasta la edad de 70 años, a fin de identificar un mayor número de pacientes con sospecha de SL (18). En referencia a la IHQ, Shia et al. sugiere la inclusión de un mayor número de marcadores en el análisis de IHQ para que su sensibilidad sea tan igual al análisis de MSI. La IHQ es una prueba de detección que presenta ventajas de disponibilidad y costos en el rastreamiento de pacientes con sospecha de SL (8).
Los criterios de Ámsterdam I y II así como las directrices de Bethesda definen el diagnóstico clínico del síndrome de Lynch y son una indicación para realizar las pruebas de análisis de mutación en los genes de MMR. Sin embargo, el presente estudio demuestra una baja sensibilidad de los Criterios de Amsterdam en nuestra población estudiada. Estos datos están en correlación con Van Lier et al. que sugiere considerar a todos los pacientes diagnosticados con cáncer colorrectal a una edad ≤ 70 años para someterse a los análisis de IHC y/o MSI para su rastreamiento (18).
Al igual que en la mayoría de los síndromes de cáncer familiar, la edad temprana de inicio y la multiplicidad de los cánceres son características en el SL. En nuestro estudio la edad media de diagnóstico fue de 60 años, edad avanzada al ser comparada con otras poblaciones de América del Sur, Argentina (44,3 años), Chile (35,7 años), Brasil (39,4 – 42,3 años) y Uruguay (35,1 años) y con las poblaciones europeas (49,7 años) (19). La diferencia en la edad puede ser debido a la falta de recursos económicos para acceder a los servicios de salud y realizar un diagnóstico temprano así como a la falta de conocimiento clínico y genético del espectro tumoral del SL por parte de los profesionales de salud.
La identificación clínica y genética del SL genera un gran impacto en la salud pública de nuestro país. La determinación de la prevalencia de este síndrome en pacientes con cáncer colorrectal permite tener el conocimiento que los pacientes afectados tienen un mayor riesgo de presentar un segundo tumor y su identificación puede llevar a recomendaciones para la detección e intervención específicas para los pacientes y sus familiares en situación de riesgo (20,21).
Conclusión
Nuestro conocimiento actual sobre la influencia genética en el riesgo de cáncer colorrectal, ha permitido establecer la importancia de la historia familiar de cáncer y el establecimiento de un registro de tumores hereditarios en el centro de salud.
Esta información nos permitirá hacer un seguimiento aquellos pacientes que se sospecha tienen un alto riesgo de padecer cáncer colorrectal hereditario con la finalidad de realizar un diagnóstico temprano y un tratamiento personalizado acompanado de un aconsejamiento genetico.
Sin embargo aunque en nuestro medio no existe un consenso sobre el método correcto para la obtención de información sobre la historia familiar de cáncer y su uso como herramienta de rastreamiento, es de gran responsabilidad de parte de los profesionales de salud identificar estos pacientes de alto riesgo y derivarlos a los servicios de genética de los centros asistenciales. El presente estudio molecular de Sindrome de Lynch demuestra la importancia del análisis molecular en familias con sospecha de cáncer colorrectal hereditario a fin de ofrecer posibilidades de vigilancia y seguimiento que han demostrado reducir la morbilidad y la mortalidad del cáncer colorrectal así como contribuir a la caracterización a nivel genética y clínica de este tipo de cáncer en la población peruana.
Conflictos de interés: Los autores declaran no tener conflictos de interés.
BIBLIOGRAFÍA
1. Balmaña J, Castells A, Cervantes A; ESMO Guidelines Working Group. Familial colorectal cancer risk: ESMO Clinical Practice Guidelines . Ann Oncol. 2010 May;21 Suppl 5:v78-81. doi: 10.1093/annonc/mdq169.
2. Lindor NM, Rabe K, Petersen GM, Haile R, Casey G, Baron J, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X . JAMA. 2005 Apr 27;293(16):1979-85..
3. Plazzer JP, Sijmons RH, Woods MO, Peltomäki P, Thompson B, Den Dunnen JT, Macrae F. The InSiGHT database: utilizing 100 years of insights into Lynch syndrome. Fam Cancer. 2013 Jun;12(2):175-80. doi: 10.1007/s10689-013-9616-0.
4. Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC) . Dis Colon Rectum. 1991 May;34(5):424-5.
5. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC . Gastroenterology. 1999 Jun;116(6):1453-6.
6. Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines . J Natl Cancer Inst. 1997 Dec 3;89(23):1758-62
7. Wielandt AM, Zárate AJ, Hurtado C, Orellana P, Alvarez K, Pinto E, et al. [ Lynch syndrome: selection of families by microsatellite instability and immunohistochemistry ]. Rev Med Chil. 2012 Sep;140(9):1132-9. doi: 10.4067/S003498872012000900005. [Article in Spanish]
8. Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry . J Mol Diagn. 2008 Jul;10(4):293-300. doi: 10.2353/jmoldx.2008.080031.
9. Valentin MD, da Silva FC, dos Santos EM, Lisboa BG, de Oliveira LP, Ferreira Fde O, ET al. Characterization of germline mutations of MLH1 and MSH2 in unrelated south American suspected Lynch syndrome individuals . Fam Cancer. 2011 Dec;10(4):641-7. doi: 10.1007/s10689-011-9461-y.
10. Zhang L. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part II. The utility of microsatellite instability testing. J Mol Diagn. 2008 Jul;10(4):301-7. doi: 10.2353/ jmoldx.2008.080062
11. Douillard JY. Microsatellite instability and mismatch repair genes in colorectal cancer: useful tools for managing patients and counseling their relatives. Clin Colorectal Cancer. 2010 Oct;9(4):193-4. doi:10.3816/CCC.2010.n.028
12. Rangel MC, Da Silva SD, Pereira NC, Dominguez MV. Essentials of Molecular Biology in Cancer Research. Applied Cancer Research. 2008 v.28, p. 2-10.
13. Dominguez-Valentin M, Nilbert M, Wernhoff P, López-Köstner F, Vaccaro C, Sarroca C, et al. Mutation spectrum in South American Lynch syndrome families . Hered Cancer Clin Pract. 2013 Dec 18;11(1):18. doi: 10.1186/1897-428711-18.
14. Koehler-Santos P, Izetti P, Abud J, Pitroski CE, Cossio SL, Camey SA, et al. Identification of patients at-risk for Lynch syndrome in a hospital-based colorectal surgery clinic . World J Gastroenterol. 2011 Feb 14;17(6):766-73. doi: 10.3748/ wjg.v17.i6.766.
15. Vaccaro CA, Carrozzo JE, Mocetti E, Berho M, Valdemoros P, Mullen E, et al. [ Immunohistochemical expression and microsatellite instability in Lynch syndrome ]. Medicina (B Aires). 2007;67(3):274-8.
16. Li D, Hu F, Wang F, Cui B, Dong X, Zhang W, et al. Prevalence of pathological germline mutations of hMLH1 and hMSH2 genes in colorectal cancer . PLoS One. 2013;8(3):e51240. doi: 10.1371/journal.pone.0051240.
17. Boland CR. Clinical uses of microsatellite instability testing in colorectal cancer: an ongoing challenge . J Clin Oncol. 2007 Mar 1;25(7):754-6.
18. van Lier MG, Leenen CH, Wagner A, Ramsoekh D, Dubbink HJ, van den Ouweland AM, et al. Yield of routine molecular analyses in colorectal cancer patients ≤70 years to detect underlying Lynch syndrome . J Pathol. 2012 Apr;226(5):764-74. doi: 10.1002/path.3963.
19. Dominguez-Valentin M, Therkildsen C, Da Silva S, Nilbert M. Familial colorectal cancer type X: genetic profiles and phenotypic features . Mod Pathol. 2014 Apr 18. doi: 10.1038/ modpathol.2014.49.
20. Castells A, Marzo-Castillejo M, Mascort JJ, Amador FJ, Andreu M, Bellas B, et al. [ Clinical practice guideline. Prevention of colorectal cancer. 2009 update. Asociación Española de Gastroenterología ]. Gastroenterol Hepatol. 2009 Dec;32(10):717.e1-58. [Article in Spanish]
21. Nique C, Sanchez F, Wernhoff P, Dominguez-Valentin M. Identificacion de cáncer colorrectal hereditario: Sindrome de Lynch. Rev cuerpo méd HNAAA. 2014;7(1).
Correspondencia: Mev Dominguez Valentin, PhD Department of Biomedicine, University of Bergen, 5009 Bergen, Norway.
E-mail: mev_dv@yahoo.com
Recibido: 04-10-2014
Aprobado: 11-11-2014

