










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Gastroenterología del Perú
Print version ISSN 1022-5129
Rev. gastroenterol. Perú vol.35 no.4 Lima Oct. 2015
REPORTE DE CASOS
Enfermedad de Wilson: forma hepática
Wilson disease: liver form
Luis Guerra Montero1, Félix Ortega Álvarez1, Julia Sumire Umeres2, Jaime Cok García3
1 Unidad de Trasplante de Órganos y Tejidos, Hospital Nacional Ramiro Priale Priale. Huancayo, Perú.
2 Servicio de Anatomía Patológica, Hospital Nacional Guillermo Almenara Irigoyen. Lima, Perú.
3 Servicio de Anatomía Patológica, Hospital Nacional Cayetano Heredia. Lima, Perú.
RESUMEN
La enfermedad de Wilson (EW) es un trastorno del metabolismo del cobre que se hereda de forma autosómica recesiva, lo cual produce acumulación tóxica del cobre principalmente en el hígado y el cerebro, en general tiene dos formas de presentación, la hepática en edades tempranas y la neurológica en edades más tardías. Se presenta el caso de una paciente mujer de 21 años diagnosticada de EW en su forma hepática en estadio cirrosis que debutó con un síndrome ascítico edematoso sin ninguna manifestación neurológica a pesar de su edad. En sus estudios de laboratorio presentó descenso de la ceruloplasmina sérica y cupruria elevada en 24 horas, datos característicos de esta enfermedad. Aunque la EW no es una enfermedad común debe ser sospechada en toda hepatopatía crónica de etiología no determinada con marcadores virales y de autoinmunidad negativos con o sin manifestaciones neurológicas ya que su reconocimiento temprano e inicio del tratamiento con quelantes del cobre principalmente conlleva a una mejora sustancial del pronóstico de vida de estos pacientes.
Palabras clave: Enfermedad de Wilson; Cirrosis hepática; Ceruloplasmina (fuente: DeCS BIREME).
ABSTRACT
Wilson disease (WD) is a disorder of copper metabolism that is inherited as an autosomal recessive, which produces toxic copper accumulation mainly in the liver and brain, in general has two ways presentation, liver at early ages and neurological in later ages. We present the case of a female patient of 21 years diagnosed of WD in liver cirrhosis that started with an edematous ascites without any neurological symptoms despite the age. Their laboratory studies showed decrease in serum ceruloplasmin and high cupruria within 24 hours of the disease , characteristic data of WD. Although WD is not a common disease should be suspected in all chronic liver disease of unknown etiology with negative viral markers and autoimmunity with or without neurological manifestations as soon as posible and starting treatment with copper chelating mainly leads to a substantial improvement the prognosis of these patients.
Key words: Wilson Disease; Liver cirrhosis; Ceruloplasmin (source: MeSH NLM).
INTRODUCCIÓN
La enfermedad de Wilson (EW), es una enfermedad autosómica recesiva causada por mutaciones en el gen ATP7B (localizado en el cromosoma 13) que codifica la proteína ATP7B que es esencial para el transporte y la excreción de exceso de cobre en el hígado, ya que incorporaelcobrelibreenlamoléculaapoceruloplasmina, que a su vez transporta el exceso de metal a las vesículas excretores de los conductos biliares (1-3). La falta de incorporación de cobre en la apoceruloplasmina produce descenso de la ceruloplasmina sérica, lo cual no tiene significación clínica (2). Cuando la gran cantidad de cobre rebasa la excreción hepática se libera grandes cantidades de cobre a la circulación general que se deposita en el cerebro, otros órganos y se elimina por las vías urinarias (4). Sus principales manifestaciones clínicas van a depender del órgano más afectado ya sea insuficiencia o cirrosis hepática, manifestaciones neurológicas o síndrome psiquiátrico (1) aunque también se han observado efectos tóxicos en el hueso, la retina, el riñón y tejidos hematológicos (3).
CASO CLÍNICO
Noviembre de 2013
Paciente femenina de 21 años de edad, estudiante procedente de La Merced (Junín) sin antecedentes familiares a destacar, dentro de sus antecedentes personales, no presenta enfermedades autoinmunes, ha sido sometida a una cirugía abdominal por una tumoración gastropancreática (probablemente un GIST) en otro hospital motivo por el cual recibió varias transfusiones sanguíneas, no ingesta de bebidas alcohólicas, medicamentos o hierbas hepatotóxicas, como antecedentes gineco-obstétricos a destacar, menarquia a los 12 años, ciclos menstruales irregulares con amenorrea de hasta tres meses. Es referida del Hospital de La Merced por un cuadro de dos meses de evolución caracterizado por tres episodios de síndrome ascítico edematoso habiendo sido manejada con diuréticos y en una ocasión con paracentesis evacuadora con diagnóstico presuntivo de cirrosis hepática.
Al examen físico presenta ictericia de piel y mucosas, angiomas estelares a nivel de tórax anterior, distensión abdominal con matidez desplazable y esplenomegalia II/III, edemas en miembros inferiores simétricos y fríos. Sus exámenes de laboratorio mostraron: ASAT: 74 U/L (VN =<25), ALAT: 24 U/L (VN =<29), fosfatasa alcalina 468 U/L (VN = 65-195), gamma glutamil transpeptidasa 32 U/L (VN = 5-38), BT: 2,82 mg/dL (VN =<1,2), BD: 1,18 mg/dL (VN =< 0,4), proteínas totales: 7,6 g/dl (VN = 6-8,3), albúmina sérica 3,1 g/dL (VN = 3,5-5), TP 24,2 segundos (VN = 11,5-13,5) , INR: 1,78 (0,8- 1,2) y plaquetas de 104 000/ mm (VN =150.000-40.000). Hb: 8.1 (VN= 13.5- 17.5) Hto 26.2% (VN= 40-54), creatinina 0.62 mg/dl (VN = 0,7-1,3), urea 13 mg/dL (VN = 10-50) con glicemia, pruebas tiroideas normales.
Al examen oftalmológico con la lámpara de hendidura no presentó el anillo Kayser-Fleischer, el examen neurológico fue normal.
El perfil viral para hepatitis A, B, C HIV, sífilis y resto del TORSH fueron negativos. Sus estudios inmunológicos ANA, ASMA, LKM, AMA, p ANCA fueron negativos. En el proteinograma electroforético no se observó ninguna alteración.
Sus estudios metabólicos ceruloplasmina 11,2 mg/dl (VN = 20-60), cupruria 84,4 ug/24h (VN = 2-30).
En los estudios de imágenes (ecografía, TAC abdominal) se evidenció un hígado disminuido de tamaño hiperecogénico con bordes irregulares, moderado líquido libre, ausencia de páncreas y esplenomegalia. En su gastroscopía no se observó várices esofágicas, anastomosis gastroyeyunal permeable sin alteraciones.
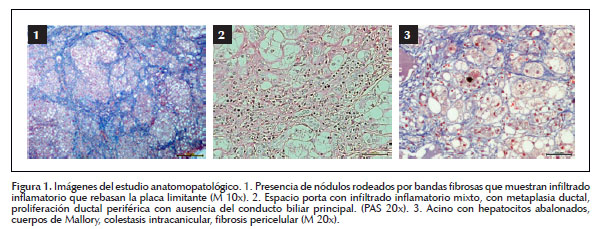
La biopsia hepática (Figura 1) mostró una arquitectura hepática distorsionada por nódulos regenerativos rodeados de bandas fibrosas con infiltrado inflamatorio mixto, ocasionales células plasmáticas y eosinófilos que rebasan la placa limitante, metaplasia ductal y proliferación ductal periférica con ausencia del conducto biliar principal en el 50% de los espacios portas, en el acino se evidenció esteatosis macrovesicular, colestasis intracanicular e infiltrado mixto (no se realizó la tinción con orceina o rodamina por no tener disponibilidad) concluyéndose cirrosis hepática secundaria a NASH o EW.
El estudio de histocompatibilidad informó HLA A2 A11, B35, B60, DR9, DR13.
Se hace el diagnóstico de cirrosis hepática por EW estratificada como un Child Pugh B (8 puntos) y MELD 17 complicada con un síndrome ascítico edematoso e ictérico además de anemia y trombocitopenia secundaria a la misma. Se inició tratamiento con espironolactona 200 mg/día furosemida 80 mg/día y sulfato ferroso 120 mg/día, D penicilamina 250 mg/día inicialmente hasta llegar a 750 mg/día, piridoxina 50 mg/día .
Enero y marzo del 2014
La paciente presentó 2 episodios de encefalopatía grado II/III siendo manejada con lactulosa VO y enemas (250 cc lactulosa más 750 cc suero fisiológico) además de metronidazol 1000 mg/día inicialmente luego con rifaximina 1200 mg/día.
La familia y paciente refirieron que en estos episodios suspendieron la medicación pues no estaba disponible en la farmacia del hospital.
Abril del 2014 Por decisión tomada en la reunión nacional de la comisión de la gerencia de procura y trasplantes se refiere a la paciente al HNGAI debido a las dificultades quirúrgicas que puede presentar, si es que el trasplante hepático se realizaría en el HNRPP dado el antecedente de gastrectomía parcial y pancreatectomía que tuvo la paciente.
Al momento de la referencia la paciente presentaba ictericia de piel y mucosas, leve ascitis y leve mejoría de su perfil hepático (ASAT: 46 U/L, ALAT: 29 U/L, fosfatasa alcalina: 245 U/L, gamma glutamil transpeptidasa: 59 U/L, BT: 5,06 mg/dL BD: 2,06 mg/ dL) y renal (creatinina 0,68 mg/dL, urea 14 mg/dL) persistiendo con prolongación del TP: 26, INR: 1,9 plaquetopenia y anemia. Con un Child Pugh C (10) MELD 20.
Su ceruloplasmina 7,9 mg/dL y cupruria 197,6 ug/24 h datos característicos de la respuesta al tratamiento.
DISCUSIÓN
La EW tiene una prevalencia de aproximadamente 1 en 30 000 - 100 000 individuos y la frecuencia de los portadores de las mutaciones del gen ATP7B es aproximadamente de 1 en 90 a 150 (5) la relación entre hombres y mujeres es controversial en los diversos estudios revisados (1). Las manifestaciones clínicas de la EW son muy diversas, pero en general en la primera década de vida hay mayor frecuencia de manifestaciones hepáticas y después de los 20 años el 75% presentan manifestaciones neurológicas y 25% manifestaciones tanto hepáticas como neuropsiquiátricas (1).
El espectro clínico de la forma hepática es muy variable y va desde hipertransaminasemias asintomáticas hasta falla hepática fulminante, pasando por episodios transitorios de ictericia debido a la hemólisis o formas de hepatitis aguda o crónica simulando hepatitis virales o autoinmunes o formas de cirrosis compensada o descompensada (6) como es el caso de nuestra paciente que debutó con un síndrome ascítico edematoso como manifestación de una hepatopatía crónica y a pesar de sus 21 años no presentaba ninguna manifestación neuropsiquiátrica.
Dentro de las manifestaciones neuropsiquiátricas se observan con mayor frecuencia temblor asimétrico de las manos (temblor postural), temblor intencional del tronco y cabeza, con menor frecuencia rigidez, distonía, bradicinesia, disartria y síntomas apráxicos, las convulsiones cerebrales ocurren con escasa frecuencia, pero más frecuentes que en la población general (4,7). Hasta un tercio de los pacientes pueden presentar inicialmente síntomas psiquiátricos como déficit de atención, depresión, cambios de humor y en casos extremos, síntomas psicóticos (7). Al examen físico un signo característico es el anillo de Kayser-Fleischer que son los depósitos granulares de cobre en la superficie interna de la córnea (membrana Descemet), se observan en el 95% de los pacientes que se presentan con síntomas neurológicos y en el 50% de los pacientes que no presentas estos síntomas (4).
En general el diagnóstico se realiza en función a criterios clínicos, analíticos (ceruloplasmina en suero < 20 mg/dL, alta concentración de cobre urinario en 24 horas > 100 mg, anemia hemolítica con Coombs negativa en caso de falla hepática fulminante) (1,6,8) e histológicos (concentración de cu hepático > 250 ug/g) (8).
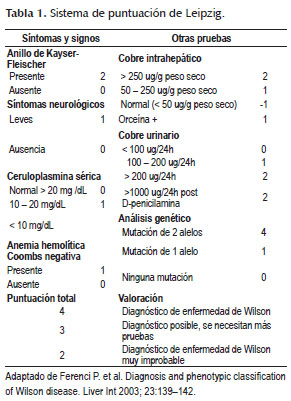
Sin embargo dado que existe una gran variabilidad de la EW a menudo su diagnóstico es difícil y a veces tardío por ello se han diseñados sistemas de puntuación útiles (Tabla 1) para hacer el diagnóstico precoz de esta entidad como la de Leipzig (9) que agrega hallazgos oftalmológicos como el anillo de Kayser-Fleischer y los síntomas neurológicos ya descritos (8)
Desde el punto de vista histológico cabe mencionar que la EW es una gran simuladora ya que no tiene datos específicos, por lo cual se puede encontrar apoptosis, esteatosis, cuerpos de Mallory como se ven en casos de NASH, nodularidad difusa, puentes fibrosos hallazgos de cualquier caso de cirrosis, con tinciones especiales como orceína o rodamina se puede hallar gránulos intrahepáticos de cobre que sin embargo no son específicas pues en los estadios iniciales de la enfermedad pueden estar ausentes (5,6). La biopsia hepática se reserva para aquellos pacientes en los cuales los signos clínicos y las pruebas no invasivas no permiten realizar un diagnóstico definitivo o existe gran sospecha de otra enfermedad hepática (5,6). Otro punto a destacar es que la severidad de las lesiones no se correlacionan con los datos de laboratorio como ocurre en la mayoría de las hepatopatías.
Algunos autores consideran concentraciones mayores de 250 ug/g cobre hepático como el gold standar para el diagnóstico de la EW (8) sin embargo, se tiene que recordar que las concentración de cobre en el hígado no tiene distribución homogénea por lo tanto la biopsia hepática tiene que ser amplia para evitar los falsos negativos además que el cobre hepático también puede estar aumentada en otras patologías colestáticas (8). Nuestra paciente presentó clínica de cirrosis, ceruloplasmina sérica disminuida cupruria elevada e histología sugestiva de EW por lo cual hicimos el diagnóstico de EW, el mismo que se confirmó totalmente con la respuesta al tratamiento quelante.
Recientemente se han introducido pruebas genéticas que fundamentalmente quedan reservadas para el cribado familiar de los casos índices y para aquellos pacientes en los que el diagnóstico es difícil de establecer por criterios clínicos y analíticos (9).
Los tratamientos existentes y empleadas en la EW son los agentes quelantes (D-penicilamina, trientina, tetratiomolibdato) y sales de zinc, en general el enfoque para el tratamiento depende de si el paciente está asintomático o sintomático y también en la principal manifestación de los síntomas (neurológicas y/o hepáticas) (1,5,6,8-10). La D-penicilamina parece más efectiva en la terapia inicial y de mantenimiento de la forma hepática, las sales de zinc en el mantenimiento de la misma y la tetratiomolibdato en la forma neurológica (4). Los pacientes con insuficiencia hepática aguda o con cirrosis descompensada que no responde al tratamiento de quelación deben ser incluidos en la lista de espera para trasplante hepático (4).
La insuficiencia hepática aguda por EW representa menos del > 5% de los casos y de estos un alto porcentaje serán fatales si es que no se someten a un trasplante hepático. Según el score pronóstico del grupo de la King’s College (Tabla 2) (basados en valores de bilirrubina, AST, INR, albumina y recuento de glóbulos blancos) los pacientes que tengan un puntaje >11 no sobrevivirán si no se realizan un trasplante (11).
Adaptado de Petrasek J. et al. Revised Kings College score for liver transplantation in adult patients with Wilsons disease. Liver Transpl 2007;13:55-61.
A nuestra paciente se le inicio tratamiento con D-penicilamina con respuesta favorable pero por el estado avanzado de su hepatopatía y que además seguía presentando descompensaciones fue incluida en lista de espera de trasplante hepático.
Conclusión
La EW es un trastorno hereditario autosómico recesivo del metabolismo del cobre que produce daño y síntomas hepático o neuropsiquiatricos de grado variable se produce por una mutación del gen ATP7B localizado en el cromosoma 13 que evita la excreción biliar del cobre, su presentación hepática abarca todo el espectro clínico de la hepatología su reconocimiento precoz mediante exploración clínica, bioquímica o histológica y el inicio temprano del tratamiento con quelantes o sales de zinc son esenciales para el resultado favorable y el pronóstico, en último caso se plantea al trasplante hepático como terapia definitiva si es que no hay respuesta al tratamiento farmacológico o cuando se presenta en forma de insuficiencia hepática aguda.
Conflicto de intereses: Los autores declaran no tener ningún conflicto de intereses.
BIBLIOGRAFÍA
1. Mansoor S, Naveed AK, Majeed A. Analysis of clinical and biochemical spectrum of Wilson disease patients. Indian J Pathol Microbiol. 2012;55(3):365-9. doi: 10.4103/0377- 4929.101746. [ Links ]
2. Dhawan A, Taylor RM, Cheeseman P, De Silva P, Katsiyiannakis L, Mieli-Vergani G. Wilson’s disease in children: 37-year experience and revised King’s score for liver transplantation. Liver Transpl. 2005;11(4):441-8.
3. Kleine RT, Mendes R, Pugliese R, Miura I, Danesi V, Porta G. Wilson’s disease: an analysis of 28 Brazilian children. Clinics (Sao Paulo). 2012;67(3):231-5.
4. Huster D, Leonhardt K, Môssner J. Wilson disease - update on pathophysiology and management. Česká a slovenská gastroenterologie a hepatologie. 2008;62(4):220-8. [ Links ]
5. European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol. 2012;56(3):671-85. doi: 10.1016/j.jhep.2011.11.007.
6. Ochoa Palominos A, Ibáñez Samaniego L, Catalina Rodríguez MV, Pajares Díaz J, Clemente Ricote G. Enfermedad de Wilson: espectro clínico de la enfermedad hepática. Gastroenterol Hepatol. 2013;36(2):86-91. doi: 10.1016/j. gastrohep.2012.07.009. [ Links ]
7. Castañeda MA, Ubilluz R, Avalos C, Escalante D, Nicoll J. Enfermedad de Wilson: Forma Neuropsiquiátrica dominante presentación de un caso y su interpretación fisiopatológica basada en resonancia magnética del encéfalo. Rev Gastroenterol Peru. 2002;22(1):74-80. [ Links ]
8. Roberts EA1, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47(6):2089-111. doi: 10.1002/hep.22261. [ Links ]
9. Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23(3):139-42. [ Links ]
10. Ostapowicz G, Fontana RJ, Schiodt FV, Larson A, Davern TJ, Han SH, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137(12):947-54. [ Links ]
11. Petrasek J, Jirsa M, Sperl J, Kozak L, Taimr P, Spicak J, et al. Revised King’s College score for liver transplantation in adult patients with Wilson’s disease. Liver Transpl. 2007;13(1):55-61.
Correspondencia: Luis Justino Guerra Montero. E-mail: guerramontero@gmail.com
Recibido: 02-10-2014
Aprobado: 06-07-2015
