










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Gastroenterología del Perú
versión impresa ISSN 1022-5129
Rev. gastroenterol. Perú vol.36 no.1 Lima ene./mar. 2016
REPORTES DE CASO
Síndrome de Lynch variante Muir-Torre: a propósito de 2 casos
Lynch syndrome, Muir Torre variant: 2 cases
María del Carmen Castro-Mujica1a, Claudia Barletta-Carrillo1b, Marisa Acosta-Aliaga1,2c, Ximena Montenegro-Garreaud1,2c
1 Instituto Nacional de Enfermedades Neoplásicas. Lima, Perú.
2 Universidad Peruana Cayetano Heredia. Lima, Perú.
a Médico genetista, b Bióloga magíster en Biología Molecular, c Médico residente de genética
RESUMEN
El síndrome de Lynch (SL), es un síndrome genético con patrón de herencia autosómico dominante, que predispone el desarrollo de cáncer colorrectal y neoplasias extracolónicas, debido a la mutación germinal en alguno de los genes reparadores de los errores de la replicación del ADN (MLH1, MSH2, MSH6 o PMS2). El Síndrome de Muir-Torre (SMT), es una variante fenotípica del SL que predispone además a desarrollar tumores de glándulas sebáceas y queratoacantomas. Presentamos el caso de dos pacientes con SMT, con más de una neoplasia relacionada al SL, lesiones cutáneas, antecedentes familiares de cáncer y estudios de inestabilidad de microsatélites e inmunohistoquímica.
Palabras clave: Síndrome de Lynch; Síndrome de Muir-Torre; Queratoacantoma; Inestabilidad de microsatélites (fuente: DeCS BIREME).
ABSTRACT
Lynch syndrome (LS) is an autosomal-dominant inherited cancer predisposition syndrome caused by germline mutations in DNA mismatch repair genes (MLH1, MSH2, MSH6 or PMS2). Muir-Torre syndrome (MTS) is a phenotypic variant of LS that includes a predisposition to sebaceous glands tumors and keratoacanthomas. We report two patients with MTS, with more than one LS-related cancer, skin lesions, family history of cancer andmicrosatellite instability and immunohistochemistry analysis.
Key words: Lynch syndrome; Muir-Torre syndrome; Keratoacanthoma; Microsatellite instability (source: MeSH NLM).
INTRODUCCIÓN
El síndrome de Lynch (SL) (OMIM #120435) (1), es una predisposición genética al cáncer colorrectal (CCR) y neoplasias extracolónicas como el cáncer de endometrio, intestino delgado, ovario, estómago, uréter, cerebro, vía biliar, principalmente. El SL se debe a la mutación germinal en uno de los genes reparadores de los errores de la replicación del ADN (MMR) (del inglés, mismatch repair) (MLH1, MSH2, MSH6 y PMS2) que se heredan mediante un patrón autosómico dominante (AD) (2). Aproximadamente el 90% de casos es debido a mutaciones en los genes MLH1 y MSH2, seguido de MSH6 en el 7%-10% de los casos y PMS2 en menos de 5% de los casos (3). Además de los genes MMR, las deleciones germinales del gen EPCAM, el cual no es un gen reparador, causan la inactivación del gen MSH2 y son la causa del SL en el 1% de los casos (3). Las mutaciones en estos genes conllevan a una deficiencia en la expresión de las proteínas del sistema MMR causando la acumulación de errores en secuencias microsatélites a nivel tumoral, volviéndolas más largas o cortas, a lo que se conoce como inestabilidad microsatelital (4,5). Existen criterios clínico-genéticos como los de Ámsterdam II y revisados de Bethesda que permiten identificar a los casos probables de SL y a los pacientes candidatos al estudio de inestabilidad de microsatélites (IMS) y/o inmunohistoquímica (IHQ) (6,7) para posteriormente dirigir el análisis de secuenciamiento génico e identificar las mutaciones germinales causantes del SL. El estudio molecular de IMS mediante el panel Bethesda, permite evaluar y comparar los patrones de los marcadores microsatélites (BAT25, BAT26, D2S123, D5S346, D17S250) en los tejidos tumorales y sangre periférica de los pacientes, teniendo como resultado: inestabilidad alta (al menos dos marcadores microsatélites son inestables), inestabilidad baja (uno de los marcadores microsatélites es inestable) y estable (ausencia de inestabilidad en los marcadores microsatélites analizados) (2). El estudio de IHQ evalúa la expresión de las proteínas del sistema MMR (MLH1, MSH2, MSH6, PMS2) en el tejido tumoral, teniendo como resultado la presencia o ausencia de expresión de estas proteínas (2). El síndrome de Muir-Torre (MT) (OMIM #158320) (8), es considerado una variante fenotípica del SL ya que comparten características moleculares y clínicas (5), sin embargo difieren debido a que esta variante posee además un riesgo aumentado a desarrollar tumores de glándulas sebáceas (sebaceo más, adenomas y/o carcinomas sebáceos, carcinoma de células basales con diferenciación sebácea, principalmente) y queratoacantomas (9). La evaluación clínico genética de los pacientes con estas lesiones cutáneas y neoplasias viscerales a edades tempranas o con antecedentes familiares de cáncer, es necesaria para descartar la presencia del SL variante MT, solicitar los estudios moleculares correspondientes y brindar la asesoría genética al paciente y sus familiares asintomáticos en riesgo, todo esto a fin de establecer un manejo y seguimiento multidisciplinario adecuados.
Presentamos los casos de dos pacientes mujeres con SL variante MT, quienes presentaban más de una neoplasia relacionada al SL y lesiones cutáneas, historia familiar de cáncer y en quienes se les realizó el estudio de IMS e IHQ.
CASO CLÍNICO 1
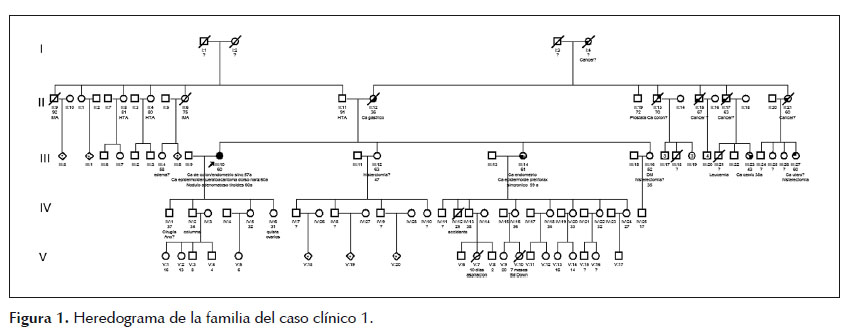
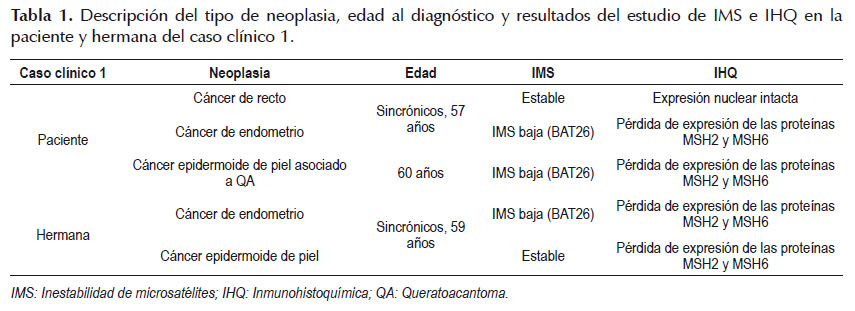
Paciente mujer de 60 años de edad, referida al consultorio de Genética por antecedente personal de adenocarcinoma moderadamente diferenciado de recto y adenocarcinoma endometroide de endometrio, sincrónicos, a los 57 años de edad y carcinoma epidermoide asociado a queratoacantoma en piel de dorso nasal a los 60 años de edad. Tras elaborar el heredograma (Figura 1), se identificó que su hermana, actualmente de 61 años de edad, fue diagnosticada de cáncer de endometrio y carcinoma epidermoide bien diferenciado de piel de tórax anterior, sincrónicos, a los 59 años de edad. Además, refirió que su madre falleció de cáncer gástrico a los 36 años de edad, cuatro tíos paternos fallecieron por cáncer desconociendo el origen primario y que sus dos hermanas restante fueron sometidas a histerectomía total sin conocer el motivo de la cirugía. Se planteó el diagnóstico de probable SL variante MT por lo que se solicitó el estudio de inestabilidad de microsatélites (IMS) e inmunohistoquímica (IHQ) en la paciente y hermana. Se brindó asesoría genética a la paciente y hermana, explicándoles los resultados (Tabla 1) y la importancia de realizar el estudio molecular principalmente del gen MSH2 y en caso de ser negativo continuar con el análisis de los genes EPCAM y MSH6 para la detección de deleciones y/o mutaciones germinales por análisis de deleción/duplicación y secuenciamiento génico completo respectivamente, además del seguimiento y control de riesgos necesarios.
CASO CLÍNICO 2
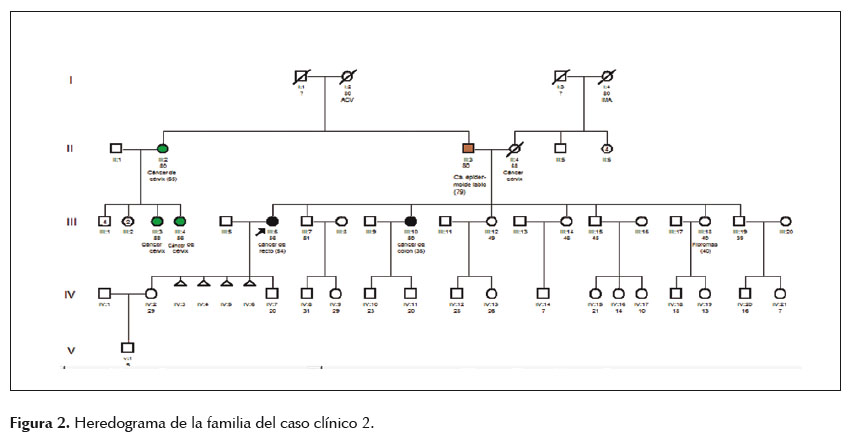
Paciente mujer de 56 años de edad, referida al consultorio de Genética por antecedente personal de adenocarcinoma tubulopapilar moderadamente diferenciado de recto a los 54 años de edad y poseer una hermana con cáncer de colon a los 35 años de edad. Tras la elaboración del heredograma (Figura 2) se confirmó el antecedente de la hermana con cáncer de colon y se añadió el antecedente del padre con cáncer epidermoide moderadamente diferenciado queratinizante de labio a los 79 años de edad. Al examen físico se evidenciaron múltiples máculas hipercrómicas en dorso de ambas manos y una lesión de aproximadamente 1,2 cm de diámetro en dorso de mano derecha, con bordes sobreelevados, eritematosos y con centro umbilicado y descamativo que impresionaba un queratoacantoma, por lo que fue referida para evaluación y biopsia excisional, confirmando el diagnóstico de queratoacantoma. Se planteó el diagnóstico de probable SL variante MT por lo que se le solicitó los estudios de IMS e IHQ. Del mismo modo al caso anterior, se brindó asesoría genética a la paciente, explicándole los resultados (Tabla 2) y la importancia de realizar el estudio molecular principalmente del gen MSH2 y en caso de ser negativo continuar con el análisis de los genes EPCAM y MSH6 para la detección de deleciones y/o mutaciones germinales por análisis de deleción/duplicación y secuenciamiento génico completo respectivamente, además del seguimiento y control de riesgos necesarios.

DISCUSIÓN
El SL es una predisposición hereditaria a desarrollar CCR y neoplasias extracolónicas como el cáncer de endometrio, principalmente, y se debe a mutaciones germinales en alguno de los genes MMR (MLH1, MSH2, MSH6 y PMS2), que se trasmiten a sus descendientes con un patrón de herencia AD (2). Todo paciente que cumpla con los criterios clínico-genéticos de sospecha de SL, debe tener una evaluación en el consultorio de genética a fin de solicitar los estudios de IMS e IHQ para posteriormente realizar la confirmación mediante el secuenciamiento de los genes MMR (5). El síndrome MT, considerado una variante fenotípica del SL, posee un riesgo aumentado a desarrollar tumores de glándulas sebáceas como la hiperplasia sebácea, epitelioma sebáceo, sebaceomas, adenomas y/o carcinomas sebáceos o múltiples queratoacantomas (QA) con diferenciación sebácea, además de alguna neoplasia visceral relacionada al SL (5,9,10). Los QA son considerados una variante del carcinoma epidermoide y se presentan principalmente en cara, dorso de manos y tórax, presentándose en el 20% de los casos de SL variante MT principalmente aquellos con diferenciación sebácea (11). Los pacientes con SL variante MT poseen antecedentes familiares de cáncer y mutaciones en línea germinal en alguno de los genes MMR, principalmente MSH2 y MLH1, y en menor frecuencia en los genes MSH6 y PMS2 (9,12-15), los cuales se heredan bajo un patrón AD, con una alta penetrancia y expresividad variable (5). El estudio de IMS e IHQ en las lesiones cutáneas y neoplasias viscerales de estos pacientes tienen como resultado, en la mayoría de casos, IMS alta y pérdida de expresión de alguna proteína MMR, principalmente MSH2 (16). Debido a que los tumores de glándulas sebáceas o queratoacantomas son infrecuentes, son considerados claves en la sospecha diagnóstica del SL variante MT (5). La mayoría de estos casos, debutan con lesiones cutáneas y posteriormente desarrollar neoplasias viscerales, siendo más frecuente el CCR u otras neoplasias extracolónicas principalmente genitourinarias (5,17), por tal motivo es necesario que todo paciente con estas lesiones cutáneas deba tener una evaluación genética para determinar si poseen criterios clínicos-genéticos de SL variante MT, identificar los antecedentes familiares, solicitar los estudios de IMS e IHQ (9,17-20) y brindar asesoría genética para el control de riesgos de neoplasias viscerales.
Los casos que presentamos, corresponden en primer lugar a una paciente con cáncer de recto, endometrio y carcinoma epidermoide asociado a queratoacantoma en piel de dorso nasal y su hermana con cáncer de endometrio y carcinoma epidermoide de piel de tórax; y el otro caso a una paciente con cáncer de recto y queratoacantoma, con antecedente familiar de hermana afectada con cáncer de colon a temprana edad. Tras el estudio de IMS e IHQ se encontró, en la mayoría de ellos, una correlación entre ambos resultados, sin embargo no todas las lesiones presentaron IMS y pérdida de expresión por IHQ. El criterio clínico-genético fue en todos los casos sugestivo de SL variante MT, por lo que un resultado "estable" tras el estudio de IMS o de "expresión intacta de las proteínas" en algunas piezas tumorales analizadas en el estudio de IHQ no puede descartar el diagnóstico. Diversos reportes muestran diferencias en los resultados del estudio de IMS entre las muestras viscerales y cutáneas de pacientes con SL variante MT (21), desde aquellos que muestran IMS alta en el 46% de neoplasias viscerales y 69% de neoplasias cutáneas (22,23) y otro donde se ha identificado IMS alta en el 71% de las lesiones cutáneas (9). Las limitaciones en nuestro trabajo es que no se cuenta con el estudio por secuenciamiento génico para la confirmación diagnóstica, sin embargo los criterios clínicos-genéticos y estudios de IMS e IHQ nos han permitido identificar a estas pacientes con SL y hacer un seguimiento del desarrollo de lesiones cutáneas para determinar que estamos frente a la variante MT.
Existen reportes, como el descrito por Tohya et al. (4) que muestra el caso de una paciente sin historia familiar de cáncer, quien desarrollo cáncer de endometrio a los 49 años y un carcinoma escamoso nasal y carcinoma sebáceo del párpado derecho, sincrónicos, a los 51 años de edad, en quien se estableció el diagnóstico de SL variante MT. Otro reporte (24), muestra el caso de una paciente con antecedentes familiares de CCR, que presentaba un quiste sebáceo en piel y carcinoma sebáceo a los 41 años y 43 años de edad, respectivamente, quien desarrolló un carcinoma de endometrio a los 49 años y múltiples carcinomas e hiperplasias sebáceas, estableciéndose el diagnóstico de SL variante MT. Tras la colonoscopía de control de riesgos se halló un CCR con ausencia de expresión de la proteína MSH2 por IHQ e IMS alta (BAT25 y BAT26 inestables). El diagnóstico fue confirmado por secuenciamiento génico tras identificar una mutación germinal heterocigota en el gen MSH2. Otros estudios como los de Ponti et al. (9) que describieron 120 casos de pacientes con lesiones de piel, de los cuáles siete habían desarrollado neoplasias viscerales, siendo cinco CCR y dos cáncer gástrico, cinco presentaron neoplasias sebáceas y dos presentaron queratoacantomas. La edad promedio fue de 54 años, la lesión sebácea fue el debut en cuatro casos y en los tres casos restantes las lesiones de piel se desarrollaron posterior a la neoplasia visceral. De los siete casos, cinco presentaron IMS alta y los resultados fueron concordantes con el estudio de IHQ, donde tres de ellos presentaron pérdida de expresión de MSH2/MSH6 y dos presentaron pérdida de MLH1. Se realizó el estudio por secuenciamiento en un caso con pérdida de expresión de MSH2/MSH6, confirmando la presencia de mutación germinal en el gen MSH2. En otro estudio de Ponti et al. (17) se describieron a cinco probandos de 57 familias que cumplían con los criterios de Amsterdam para SL y que poseían tumores de glándulas sebáceas o queratoacantomas, cuatro de ellos desarrollaron neoplasias viscerales a una edad promedio de 51,4 años, las cuáles fueron CCR, cáncer de endometrio, ovario, páncreas y mama. El estudio de IMS de las lesiones en piel se realizó en 4 de los 5 casos, encontrándose IMS alta y pérdida de expresión en IHQ. El estudio de IMS de las lesiones viscerales revelaron IMS alta en todos los casos y el estudio de IHQ mostró la pérdida de expresión MSH2/MSH6 en tres casos y pérdida de expresión de MLH1 en dos casos, siendo confirmados por secuenciamiento génico tras identificar mutaciones germinales en los genes MSH2 y MLH1 respectivamente.
Finalmente, en un reporte realizado por Nishizawa et al. (11), describieron el caso de una mujer de 56 años con antecedente de CCR y cáncer de endometrio, metacrónicos, quien presentó lesiones epiteliales desde los 46 años que incluían el carcinoma sebáceo, epitelioma sebáceo, carcinoma escamoso y tras el estudio de IHQ se identificó la perdida de expresión de MSH2 en todas las lesiones cutáneas incluyendo en el cáncer de endometrio y CCR.
El diagnóstico clínico del SL variante MT puede ser complicado debido a que requiere la coexistencia de al menos un tumor sebáceo o queratoacantoma asociado a un CCR o neoplasia extracolónica del espectro del síndrome de Lynch, además de antecedentes familiares oncológicos (2). La mayoría de veces, el SL variante MT es subdiagnosticado debido a que no se indaga adecuadamente sobre los antecedentes personales o familiares de tumores de glándulas sebáceas y/u otras neoplasias viscerales, por lo que el estudio molecular en los casos de sospecha clínica es importante para identificar a estos pacientes y realizar un seguimiento de control de riesgos en ellos y sus familiares mediante colonoscopías, ecografía transvaginal con biopsia, endoscopia alta, ecografía abdomino pélvica, estudios de función renal, cistoscopías, análisis de orina anual y examen físico en busca de lesiones cutáneas (2). Como este tipo de neoplasias cutáneas se presentan con muy baja frecuencia en la población general, podrían considerarse como un marcador para el SL variante MT y así orientar la búsqueda de neoplasias viscerales en estos pacientes (25) y evaluar los antecedentes familiares de cáncer, además de solicitar el correspondiente estudio de IMS e IHQ y brindar la asesoría genética oportuna para el control de riesgos. Esperamos que este reporte de casos contribuya en el diagnóstico y manejo de casos similares en nuestro país.
BIBLIOGRAFÍA
1. Kniffin CL. #120435 Lynch Syndrome I [Internet]. Baltimore: OMIM; 2016 [citado el 14 de junio de 2015]. Disponible en: http://omim.org/entry/120435 [ Links ]
2. Bandres F, Urioste M. Planteamientos básicos del cáncer hereditario: principales síndromes. Madrid: Fundación Tejerina; 2011. [ Links ]
3. Kohlmann W, Gruber SB. Lynch Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington; 2014 [citado el 14 de junio de 2015]. Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK1211/ [ Links ]
4. Tohya T, Ogura T, Nishi K, Nishi H, Kuriwaki K. Muir-Torre syndrome associated with endometrial carcinoma . Int J Clin Oncol. 2008;13(6):559-61. [ Links ]
5. Vaglio Giors G, Leiva MJ, Ferrario D, Volonteri V, Kowalczuk A, Vaccaro C, et al. Tumores sebáceos: ¿Tan inocentes como creemos? Síndrome de Muir-Torre . Dermatología CMQ. 2014;12(1):24-8. [ Links ]
6. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC . Gastroenterology. 1999;116(6):1453-6. [ Links ]
7. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability . J Natl Cancer Inst. 2004;96(4):261-8. [ Links ]
8. Kniffin CL. #158320 Muir-Torre Syndrome [Internet]. Baltimore: OMIM; 2016 [citado el 14 de junio de 2015]. Disponible en: http://www.omim.org/entry/158320?search=muir%20torre&highlight=torre%20muir
9. Ponti G, Losi L, Di Gregorio C, Roncucci L, Pedroni M, Scarselli A, et al. Identification of Muir-Torre syndrome among patients with sebaceous tumors and keratoacanthomas: role of clinical features, microsatellite instability, and immunohistochemistry . Cancer. 2005;103(5):1018-25. [ Links ]
10. Ponti G, Pellacani G, Seidenari S, Pollio A, Muscatello U, Tomasi A. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes . Crit Rev Oncol Hematol. 2013;85(3):239-56. [ Links ]
11. Nishizawa A, Nakanishi Y, Sasajima Y, Yamazaki N, Yamamoto A. Muir-torre syndrome with intriguing squamous lesions: a case report and review of the literature . Am J Dermatopathol. 2006;28(1):56-9. [ Links ]
12. Ponti G, Ponz de Leon M. Muir-Torre syndrome . Lancet Oncol. 2005;6(12):980-7. [ Links ]
13. Ko CJ. Muir-Torre syndrome: Facts and controversies . Clin Dermatol. 2010;28(3):324-9. [ Links ]
14. Tavakkol Z, Keller JJ, Furmanczyk PS, Bennett RL, Chien AJ. Germline mutation in MSH6 associated with multiple malignant neoplasms in a patient With Muir-Torre syndrome . J Clin Oncol. 2012;30(22):e195-8. [ Links ]
15. Kacerovska D, Cerna K, Martinek P, Grossmann P, Michal M, Ricar J, et al. MSH6 mutation in a family affected by Muir-Torre syndrome . Am J Dermatopathol. 2012;34(6):648-52. [ Links ]
16. Mathiak M, Rütten A, Mangold E, Fischer H-P, Ruzicka T, Friedl W, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test . Am J Surg Pathol. 2002;26(3):338-43. [ Links ]
17. Ponti G, Losi L, Pedroni M, Lucci-Cordisco E, Di Gregorio C, Pellacani G, et al. Value of MLH1 and MSH2 mutations in the appearance of Muir-Torre syndrome phenotype in HNPCC patients presenting sebaceous gland tumors or keratoacanthomas . J Invest Dermatol. 2006;126(10):2302-7. [ Links ]
18. Lynch HT, Fusaro RM, Lynch PM. Sebaceous skin lesions as clues to hereditary non-polyposis colorectal cancer . J Invest Dermatol. 2006;126(10):2158-9. [ Links ]
19. Hare HH, Mahendraker N, Sarwate S, Tangella K. Muir-Torre syndrome: a rare but important disorder . Cutis. 2008;82(4):252-6. [ Links ]
20. Navi D, Wadhera A, Fung MA, Fazel N. Muir-Torre syndrome . Dermatol Online J. 2006;12(5):4. [ Links ]
21. Kruse R, Rütten A, Lamberti C, Hosseiny-Malayeri HR, Wang Y, Ruelfs C, et al. Muir-Torre phenotype has a frequency of DNA mismatch-repair-gene mutations similar to that in hereditary nonpolyposis colorectal cancer families defined by the Amsterdam criteria . Am J Hum Genet. 1998;63(1):63-70. [ Links ]
22. Entius MM, Keller JJ, Drillenburg P, Kuypers KC, Giardiello FM, Offerhaus GJ. Microsatellite instability and expression of hMLH-1 and hMSH-2 in sebaceous gland carcinomas as markers for Muir-Torre syndrome . Clin Cancer Res. 2000;6(5):1784-9. [ Links ]
23. Popnikolov NK, Gatalica Z, Colome-Grimmer MI, Sánchez RL. Loss of mismatch repair proteins in sebaceous gland tumors . J Cutan Pathol. 2003;30(3):178-84. [ Links ]
24. Rios CA, Villalón R, Muñoz J, Acuña M, Cifuentes L. Muir-Torre syndrome: case report and molecular characterization . São Paulo Med J. 2014;132(1):61-4. [ Links ]
25. Tsalis K, Blouhos K, Vasiliadis K, Tsachalis T, Angelopoulos S, Betsis D. Sebaceous gland tumors and internal malignancy in the context of Muir-Torre syndrome. A case report and review of the literature . World J Surg Oncol. 2006;4:8. [ Links ]
Correspondencia: María del Carmen Castro Mujica
Equipo Funcional de Genética y Biología Molecular
Instituto Nacional de Enfermedades Neoplásicas
Av. Angamos Este 2520. Lima 34. Perú.
E-mail: mcastro@inen.sld.pe
Recibido: 08-07-2015
Aprobado: 04-10-2015
