










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Gastroenterología del Perú
Print version ISSN 1022-5129
Rev. gastroenterol. Perú vol.36 no.2 Lima Apr./Jun. 2016
REPORTES DE CASOS
Síndrome de Peutz- Jeghers. Presentación de cinco casos
Peutz Jeghers syndrome. Report of five cases
Bertha Idrogo Regalado1a, Oscar Frisancho Velarde1b
1 Departamento Enfermedades del Aparato Digestivo, Hospital Nacional Edgardo Rebagliati Martins. Lima, Perú.
a Médico residente; b Médico asistente
RESUMEN
El Síndrome de Peutz Jeghers (SPJ) es caracterizado por pólipos hamartomatosos gastrointestinales y pigmentación mucocutánea característica, tiene alto riesgo de resecciones intestinales debido a isquemia secundaria e intususcepción. El riesgo de cáncer digestivo es nueve veces más que en la población general. Reportamos cinco pacientes con diagnóstico de SPJ, tres casos presentaron intususcepción intestinal, un caso hemorragia digestiva alta y el quinto caso falleció con neoplasia pancreática.
Palabras clave: Neoplasias gástricas; Pólipos del colon; Poliposis intestinal (fuente: DeCS BIREME).
ABSTRACT
The Peutz Jeghers Syndrome (PJS) is characterized by gastrointestinal hamartomatous polyps and mucocutaneous pigmentation, are at high risk of bowel resections because ischemia secondary to intussusception. The risk of gastrointestinal cancer is nine more than the general population. We report five patients diagnosed with SPG, four had intestinal intussusception, one upper gastrointestinal bleeding and one died with pancreatic neoplasia.
Key words: Stomach neoplasms; Colonic polyps; Intestinal polyposis (source: MeSH NLM).
INTRODUCCIÓN
El SPJ es caracterizado por pólipos hamartomatosos gastrointestinales y pigmentación mucocutánea, tiene alto riesgo de resecciones intestinales debido a isquemia por invaginación intestinal. El riesgo de cáncer digestivo es nueve más que en la población general (1).
Los pólipos hamartomatosos se ubican a lo largo de todo el tracto gastrointestinal siendo más frecuente en colon (60%), estómago (25-50%) e intestino delgado (20-40%); pueden ser pediculados o sésiles, tienen tamaño variable, de mayor dimensión en el intestino delgado, el riesgo acumulativo de desarrollar cualquier tipo de tumor maligno en la región pancreatobiliar, incluyendo páncreas, vía biliar distal, o cáncer ampular, es tan alta como 29% a la edad de 70 años (2).
La pigmentación mucocutánea es de tipo melánica, en forma de máculas pequeñas (lentigines) de 1 a 5 mm alrededor de la boca, ojos, ano, dedos de manos y pies; las lesiones mucosas persisten largos años, las cutáneas pueden remitir con el tiempo.
Reportamos cinco pacientes con diagnóstico de SPJ, tres presentaron intususcepción intestinal, uno hemorragia digestiva alta y uno falleció con neoplasia pancreática.
CASOS CLÍNICOS
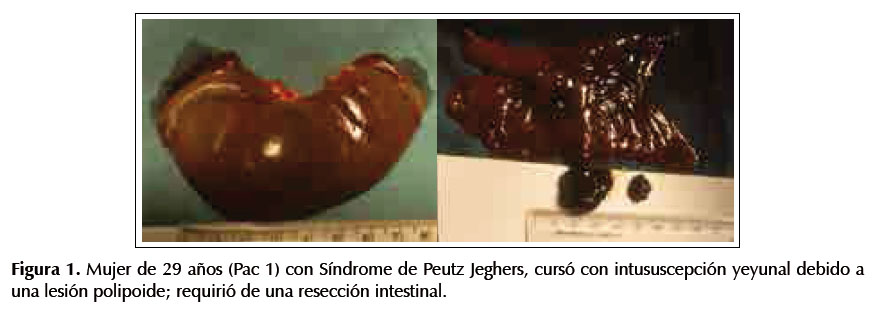
Paciente 1.-Mujer de 29 años, con manchas hipercrómicas en labios y mucosa oral (tienen dos hermanas con melanosis similar) y diagnóstico endoscópico de pólipos gástricos y colónicos múltiples. Cursó con intususcepción yeyunal debido a un pólipo grande. Ver Figura 1.
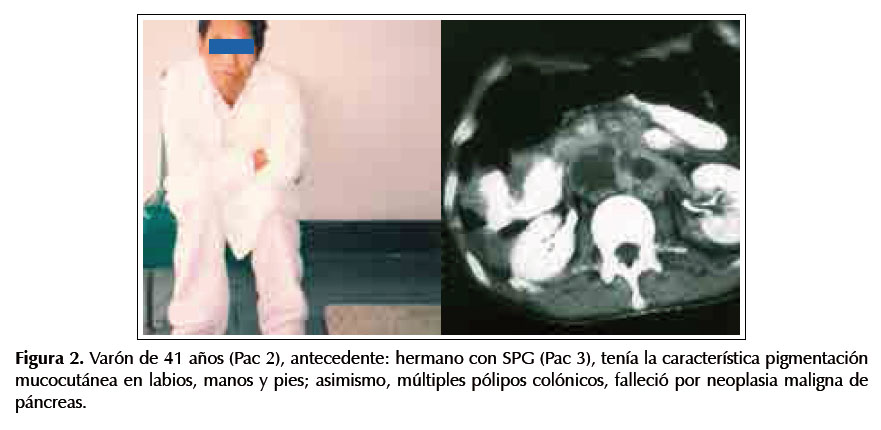
Paciente 2.-Varón de 41 años, antecedente hermano con SPJ (Pac 3), tiene melanosis en labios, manos y pies; asimismo, múltiples pólipos colónicos. Falleció por neoplasia maligna de páncreas. Ver Figura 2.
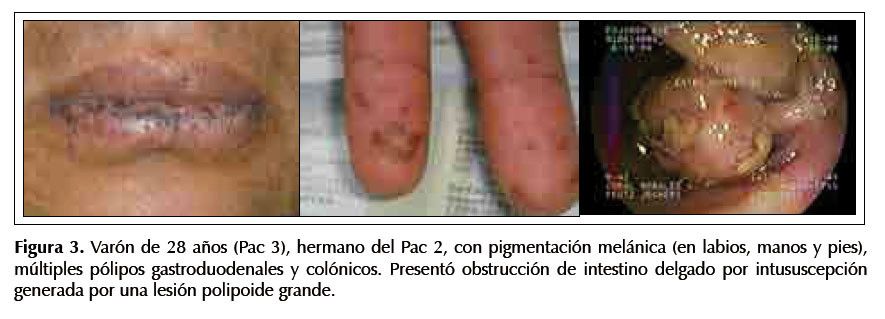
Paciente 3.-Varón de 28 años, hermano del Pac 2, tiene melanosis en labios, manos y pies (tiene una hija con melanosis similar) y múltiples pólipos gastroduodenales y colónicos. Cursó con obstrucción de intestino delgado por intususcepción. Ver Figura 3.
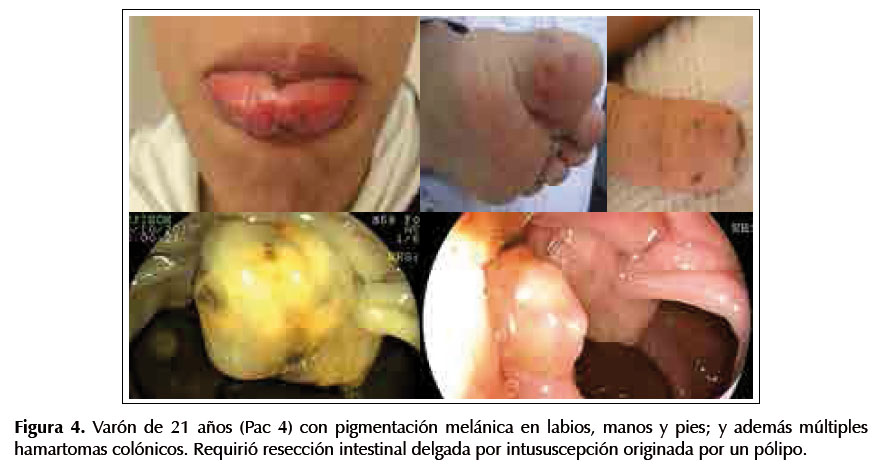
Paciente 4.-Varón de 21 años, con melanosis en labios, manos y pies (su abuela paterna tiene lesiones similares) y múltiples hamartomas colónicos. Requirió resección intestinal delgada por intususcepción originada por un pólipo. Ver Figura 4.
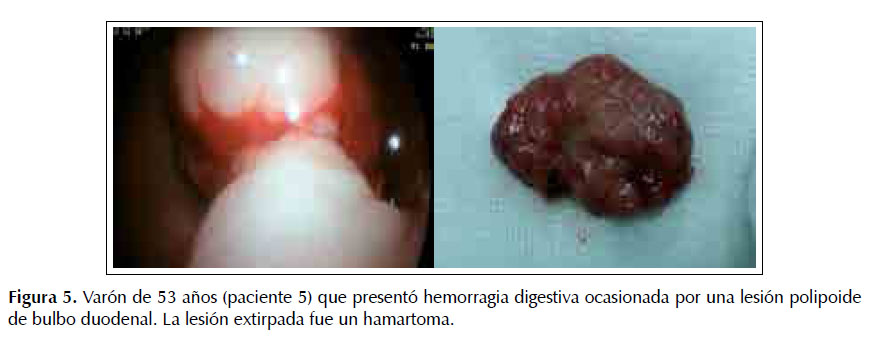
Paciente 5.-Varón de 51 años, con melanosis en palma de manos, presentó hemorragia digestiva alta y anemia severa (Hb 4,5 gr/dl). No tenía estudios endoscópicos anteriores; su anterior episodio sangrante fue hace dos meses. En la endoscopia digestiva alta se detectó que el sangrado provenía de un "pólipo" pediculado de 30x40 mm en cara anterior de bulbo duodenal; la lesión fue extirpada con asa de electrocoagulación. El estudio de anatomía patológica demostró un hamartoma duodenal. Ver Figura 5.
DISCUSIÓN
El Síndrome de Peutz-Jeghers es un síndrome autosómico dominante caracterizado por múltiples pólipos hamartomatosos en el tracto gastrointestinal, pigmentación mucocutánea e incremento de riesgo de tener cáncer gastrointestinal. Un hamartoma es una hiperplasia localizada de células epiteliales intestinales maduras que se asienta sobre un núcleo de submucosa (3-5).
Es una entidad rara con prevalencia estimada de 1:8000 a 1:200 000, con igual afectación en hombres y mujeres (6).
Está asociado con la mutación germinal en el gen STK11 (LKB1) que codifica una serinatreonina quinasa mapeado en el cromosoma 19p (7,8).
Existen dos tipos principales de manifestaciones clínicas del SPJ (9):
1.- Pigmentación mucocutánea en forma de máculas y múltiples pólipos gastrointestinales. La pigmentación mucocutánea característica (manchas de melanina) están presentes en más del 95% de los pacientes, presentes en labios, región peri oral, palma de manos, mucosa oral y planta de pies.
2.- Los pólipos hamartomatosos están presentes en la mayoría de pacientes con SPJ y pueden ocurrir en cualquier lugar del tracto gastrointestinal. Los pólipos se desarrollan en la primera década de la vida y muchos pacientes son sintomáticos entre los 10 y 30 años de edad. El rango de pólipos es de 1 a más de 20 por segmento de intestino.
El riesgo de cáncer gastrointestinal oscila entre 37 y 93% con un promedio de edad de 42 años al hacer el diagnóstico (1).
Los sitios más comunes de desarrollar tumores malignos del tracto gastrointestinal en el Síndrome de Peutz Jeghers son el colon y el páncreas y los sitios más comunes de tumores extra intestinales son las mamas. Las mujeres también tienen un mayor riesgo de tumores cervicales incluyendo adenoma maligno de cuello de útero y los tumores ováricos benignos conocidos como tumores "sexcord" con túbulos anulares. Los hombres con SPJ tienen un mayor riesgo de por vida de tumores testiculares de células de Sertoli (2,10).
El diagnóstico clínico de SPJ requiere la presencia de cualquiera de los siguientes:
-Dos o más pólipos con histología confirmada de Peutz-Jeghers.
- - Cualquier número de pólipos Peutz-Jeghers detectados en un individuo quien tiene historia familiar de Peutz-Jeghers o parientes cercanos.
- - Cualquier número de pólipos de Peutz-Jeghers en un individuo quien tiene pigmentación mucocutánea (1,11).
Cerca del 50% de pacientes son asintomáticos hasta antes del diagnóstico, algunos pacientes presentan obstrucción intestinal causada por intususcepción u oclusión del lumen gastrointestinal causado por el pólipo, además dolor abdominal por infarto, sangrado rectal agudo o crónico causado por ulceración del pólipo. Cerca del 69% de pacientes experimentan intususcepción durante su vida, siendo más frecuente en el intestino delgado (12,13).
Pacientes con criterios clínicos de Peutz-Jeghers, deben someterse a un test genético de mutación en el gen STK11. Las pruebas genéticas de un individuo que cumple con los criterios clínicos para SPJ sirve para confirmar el diagnóstico; sin embargo, la ausencia de una mutación patogénica de STK11 en una persona que cumple con los criterios clínicos para SPJ no excluye el diagnóstico de SP (14,15).
Es importante realizar despistaje en los familiares de los pacientes diagnosticados con SPG por su predisposición genética, asimismo complementar en los pacientes la existencia de pólipos en intestino delgado mediante radiografías contrastadas o enteroscopia; realizar exámenes periódicos ginecológicos a predominio de mama y urológicos, de igual manera el tratamiento con polipectomías por riesgo de neoplasia de los mismos.
De los cinco casos presentados, tres de ellos cursaron con intususcepción intestinal originada por uno de los pólipos en intestino, otro de ellos fue diagnosticado por hemorragia digestiva alta originada por laceración de pólipo duodenal observado durante endoscopia y uno de ellos presentó cáncer de páncreas, una de las neoplasias más frecuentes según menciona la bibliografía.
Respecto a casos nacionales con SPG publicados, encontramos cuatro reportes (4,14,16,17): el primer reporte fue realizado por Carlos Merino (Hospital Obrero-SSP) en 1965, el segundo reporte fue de Luis Malca
(H. Almenara-IPSS) en 1986, el tercer reporte fue de Adelina Lozano y col. (H. Loayza), y el cuarto por Juan Pinto y col. (Hospital de Policía) el 2004. Los casos fueron estudiados por dolor abdominal, anemia crónica, invaginación intestinal, entre otros; el paciente reportado por Pinto (4) tenía cáncer de colon. Lozano (14) describe dos pacientes hermanos (hombre y mujer) con crisis suboclusivas a repetición, su madre fallecida por cáncer de colon; además, la paciente tenía una hija de un año y medio con melanosis mucocutánea asintomática.
BIBLIOGRAFÍA
1. Calva D, Howe JR. Hamartomatous polyposis syndromes. Surg Clin North Am. 2008;88(4):779-817. [ Links ]
2. Korsse SE, Harinck F, van Lier MG, Biermann K, Offerhaus GJ, Krak N, et al. Pancreatic cancer risk in Peutz-Jeghers syndrome patients: a large cohort study and implications for surveillance . J Med Genet. 2013;50(1):59-64. [ Links ]
3. Edwards DP, Khosraviani K, Stafferton R, Phillips RK. Long-term results of polyp clearance in the Peutz-Jeghers syndrome . Dis Colon Rectum. 2003;46(1):48-50. [ Links ]
4. Pinto Sánchez JF, Rebaza Vásquez S, Muñoz Mendoza S, Maco Cárdenas V. Síndrome de Peutz-Jeghers y adenocarcinoma de colon . Rev Gastroenterol Peru. 2004;4(4):34-6. [ Links ]
5. Harned RK, Buck JL, Sobin LH. The hamartomatous polyposis syndrome: clinical features . AJR Am J Roentgenol. 1995;164(3):565-71. [ Links ]
6. Barboza Besada E. Neoplasias intestinales. En: Tópicos Selectos en Medicina Interna. Capítulo 16. Lima: Colegio Médico del Perú; 2010. p. 238-40. [ Links ]
7. Mcgarrity TJ, Kulin HE, Zaino RJ. Peutz-Jeghers syndrome . Am J Gastroenterol. 2000;95(3):596-604. [ Links ]
8. Corredor J, Wambach J, Barnard J. Gastrointestinal polyps in children: advances in molecular genetics, diagnosis and management . J Pediatr. 2001;138(5):621-8. [ Links ]
9. Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome . Gastroenterology. 2000;119(6):1447-53 . [ Links ]
10. Srivatsa PJ, Keeney GL, Podratz KC. Disseminated cervical adenoma malignum and bilateral ovarian sex cord turmors with annular tubules associated with Peutz-Jeghers syndrome . Gynecol Oncol. 1994;53(2):256-64. [ Links ]
11. Boardman LA, Thibodeau SN, Schaid DJ, Lindor NM, McDonnell SK, Burgart LJ, et al. Increased risk for cancer in patients with Peutz Jeghers syndrome . Ann Intern Med. 1998;128(11):896-9. [ Links ]
12. Jansen M, de Leng WW, Baas AF, Myoshi H, Mathus-Vliegen L, Taketo MM, et al. Mucosal prolapse in the pathogenesis of Peutz Jeghers polyposis . Gut. 2006;55(1):1-5. [ Links ]
13. Settaf A, Mansori F, Bargach S, Saidi A. [ Peutz-Jeghers syndrome with carcinomatous degeneration of a duodenal hamartomatous polyp ]. Ann Gastroenterol Hepatol (Paris). 1990;26(7):285-8. [Article in French] [ Links ]
14. Lozano A, Valencia V, Zevallos M, Contreras R, Vargas G, Verona R. Síndrome de Peutz-Jeghers. A propósito de un reporte familiar en el Hospital Arzobispo Loayza . Rev Gastroenterol Peru. 1996;16(1):23-5. [ Links ]
15. Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, et al. Peutz –Jeghers syndrome is caused by mutations in a novel serine threonine kinase . Nat Genet. 1998;18(1):38-43. [ Links ]
16. Merino C. Poliposis intestinal asociado a melanosis cutánea. Rev Cuerpo Médico del Hospital Obrero de Lima. 1965;1:62-70. [ Links ]
17. Malca Cubas L. Síndrome de Peutz Jeghers. Rev Gastroenterol Peru. 1986;6(2):102-6. [ Links ]
Correspondencia:
Bertha Idrogo Regalado
E-mail: berthaidrogor@hotmail.com
Recibido: 26-8-2014
Aprobado: 23-6-2015
