










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Gastroenterología del Perú
versión impresa ISSN 1022-5129
Rev. gastroenterol. Perú vol.36 no.3 Lima jul./set. 2016
REPORTE DE CASO
Type 1 autoimmune pancreatitis: case scenario and review of the disease
Pancreatitis autoimmune tipo 1: caso y revisión de la enfermedad
Jean A. Donet1, Frank Czul1, Nathalie A. Peña2, Jamie S. Barkin1
1 Division of Gastroenterology, University of Miami Leonard Miller School of Medicine, Miami, FL, USA.
2 Depar tment of Medicine, University of Miami Leonard Miller School of Medicine, Miami, FL, USA.
ABSTRACT
Autoimmune pancreatitis (AIP) is an uncommon disease that represents a diagnostic challenge unless it is considered as a cause of acute pancreatitis, pancreatic exocrine insufficiency and a pancreatic mass. This entity is under diagnosed and successful medical therapy is available. In this paper, we will describe a case of a 59 year-old, Hispanic woman diagnosed with autoimmune pancreatitis, a disease previously believed to affect typically older men. We will review the definition, types, clinical manifestations, radiological features, serology, histopathological findings, treatment strategies and diagnostic criteria of autoimmune pancreatitis.
Keywords: Pancreatitis; Exocrine pancreatic insufficiency; Cholangitis, sclerosing (source: MeSH NLM).
RESUMEN
La pancreatitis autoinmune (PAI) es una enfermedad rara que se presenta como un reto diagnóstico a menos que sea considerada como causa de pancreatitis aguda, insuficiencia pancreática exocrina y masa pancreática. Es una enfermedad sub diagnosticada y existe una terapia médica satisfactoria. En este trabajo, describiremos un caso de una mujer hispana de 59 años diagnosticada de pancreatitis autoinmune, una enfermedad que se creía previamente que afectaba típicamente a hombres de avanzada edad. Revisaremos la definición, los tipos, las manifestaciones clínicas, hallazgos radiológicos, serología, hallazgos histopatológicos, estrategias de tratamiento y criterios diagnósticos de la pancreatitis autoinmune.
Palabras clave: Pancreatitis; Insuficiencia pancreática exocrina; Colangitis esclerosante (fuente: DeCS BIREME).
INTRODUCTION
Autoimmune pancreatitis (AIP) is composed of at least two disease entities. Type 1 AIP is the pancreatic manifestation of IgG4-related autoimmune diseasecharacterized by increased serum IgG4, lymphoplasmacytic infiltrates on histology, other organ involvement and dramatic response to steroid therapy. Type 2 AIP presents as an isolated organ disorder with granulocytic epithelial lesions on histology. Although the pathogenic mechanism remains unclear, multiple factors such as genetic background and abnormal immunity may be involved. Diagnosis of both types can be made using the International Consensus Diagnostic Criteria. There is consensus for initial steroid treatment; however, steroid maintenance and treatment for relapses are controversial. In the long term, approximately 10% of type 1 AIP may progressinto chronic pancreatitis. Although controversial, autoimmune pancreatitis seems to be a risk factor for pancreatic malignancy (1-3). Thus, these patients need to be followed in time as patients with chronic pancreatitis are.
CASE REPORT
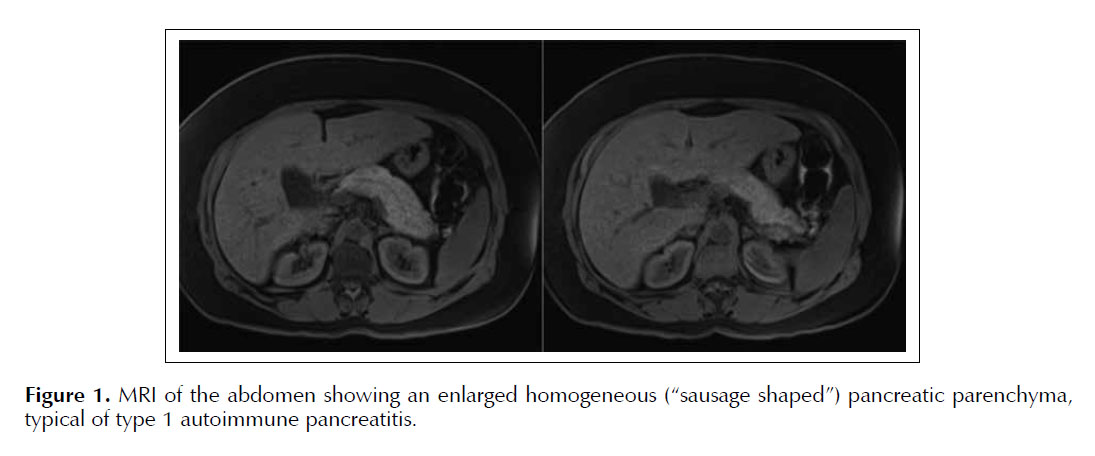
A 59 year-old Honduran womanpresented with a 3-week history of nausea, vomiting, fever, productive cough and watery diarrhea. Her past medical history was significant for acute recurrent pancreatitis ofunknown etiology. She had no history of hypertriglyceridemia, hypercalcemia or gallstones. Her family history had no history of pancreatic disease or cystic fibrosis. She denied any alcohol intake, tobaccoor drug use. Her physical exam showed crackles on the left pulmonary base, abdominal exam revealed mild periumbilical tendernessbutwas otherwise benign. Laboratory data showed leukocytosis WBC 16 400 cells/ml, normal Hb, platelet count, basic metabolic panel and liver function panel. Amylase was 163 and Lipase 644 (>3 times ULN). Ultrasound and plain CT scan of her abdomen showed a normal gallbladder without stones and an unremarkable pancreas. Nevertheless, her gastrointestinal symptoms worsenwhen herlipase levelsrose to 1553. MRI of the abdomenrevealed an enlarged homogeneous pancreatic parenchyma - sausage shaped pancreas without pancreatic duct dilation (see Figure 1) whichsuggested autoimmune pancreatitis. IgG4 level was 522 mg/dl (normal levels< 200 mg/dl). Endoscopic ultrasound was unavailable at the time and it was elected to give the patient a diagnostic / therapeutic trial of prednisone 40 mg PO daily. There was resolution of patient’s gastrointestinal symptomsand normalization of IgG4 levels. Follow-up MRI revealed resolution of the pancreatic changes. Prednisone dosage was slowly tapered over a four-month period. Patient has not had a relapse since this episode.
DISCUSSION AND REVIEW OF THE DISEASE
Classification
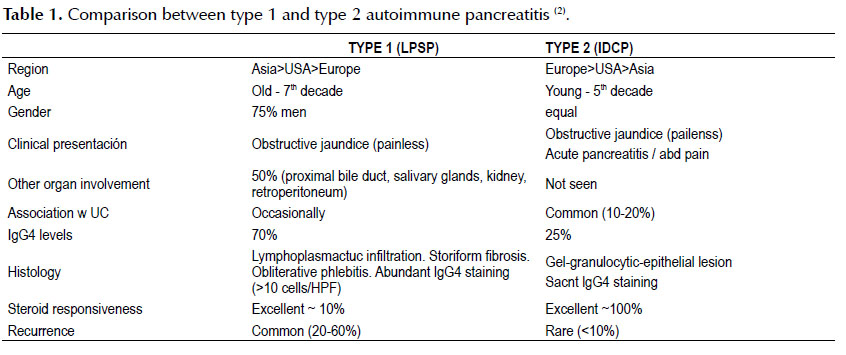
Two types of AIP have been described in the literature. Type 1 AIP also known as lymphoplasmacytic sclerosing pancreatitis (LPSP) is part of theIgG4 related diseases and therefore its clinical presentation can be divided in pancreatic and extra-pancreatic clinical manifestations. In the active phase, Type 1 AIP patients can present with obstructive jaundice, diffuse or segmental pancreatic enlargement with or without a pancreatic mass on imaging and steatorrhea or diabetes mellitus. In a more advanced state (burnt-out phase), the pancreas can contain calcifications and/or stones or have parenchymal atrophy. Pancreatic exocrine and endocrine insufficiency can also be seen. Involvement of extra-pancreatic organs is usual, themost common associated entities include sclerosing cholangitis, renal mass/tubulointerstitial nephritis, retroperitoneal fibrosis with or without ureteral obstruction and bilateral submandibular masses (Mikulicz disease) (1-3). Type 2 AIP also known as idiopathic duct central pancreatitis (IDCP) presents as an isolated pancreatic disorder with granulocytic epithelial lesions on histology. Whereas type 1 AIP where relapses are common, in type 2 AIP relapses are uncommon. Table 1 shows the main differences between type 1 and type 2 AIP.
Imaging
Magnetic resonance imaging (MRI) allows us to evaluate pancreatic anatomy for features of AIP (4). The changes can be classified into those of the parenchyma orduct. Typical parenchymal changes include diffuse enlargement of the pancreas with featureless borders, the so-called "sausage shaped pancreas" (Figure 1). A hypoattenuating capsule-like ring is also typical. A focal mass in the pancreas is an atypical radiological presentation of AIP and therefore it must be differentiated from pancreatic adenocarcinoma (5). Duct changes include narrowing of the segment of the pancreatic duct within the affected parenchyma, which is similar to that found in pancreatic cancer; however, absent or slight upstream dilatation is usually seen in AIP. Marked upstream pancreatic duct dilation is usually found resulting from pancreatic cancer. One study showed focal enlargement of the pancreas in 67% of the patients with AIP, diffuse enlargement in 33% and a hypo attenuating parenchyma in 90% of patients. The main pancreatic duct was stenotic or not visible within the parenchyma affected in all the patients. Of note, all these changes seen on CT normalized after treatment with immunosuppressors (4).
Serology
The most useful serological marker for the diagnosis of type 1 AIP is IgG4, as it is elevated in 75% of patients. IgG4 can also be elevated in other conditions that simulate AIP including pancreatic cancer, chronic pancreatitis, primary biliary cirrhosis, primary sclerosing cholangitis and Sjogren disease.
IgG4 levels can be elevated in up to 10% of patient with pancreatic adenocarcinoma, although levels are usually less than two fold the upper limit of normal. IgG4 levels above 135 mg/dl have a sensitivity of 50 to 92% and specificity higher than 90% for the diagnosis of AIP. IgG4 levels might correlate with disease activity, higher presence of extra-pancreatic manifestations and higher relapse rate (6). Combining the tumor marker CA 19.9 (<74 U/ml) and IgG4 levels (>100 mg/dl) measurement has proven to be useful to distinguish AIP from pancreatic cancer with sensitivity of 94% and specificity of 100% (7).
Histopathology
Endoscopic ultrasound (EUS) with tissue acquisition has an important diagnostic role for those patients with indeterminate findings on CT, or typical radiological findings but lack of collateral evidence (serology, extra- pancreatic involvement). EUS allows tissue acquisition via FNA or trucut biopsy. Fine needle aspiration (FNA) can be performed to obtain asample for cytology, most useful to distinguish from pancreatic cancer. FNA has the disadvantage of not preserving the pancreatic architecture andcytology can be falsely negative in up to 40% of patients withpancreatic cancer. On the other hand, a trucut biopsy obtained via EUS preserves the histological architecture and allows immunohistochemistry studies (8). Pathology remains the gold-standard test for the diagnosis of type 1 and type 2 AIP. Lymphoplasmacytic infiltrates, storiform fibrosis, obliterative phlebitis and abundant IgG4 staining are typical of type 1 AIP. The presence of granulocytic epithelial lesions is characteristic of type 2 AIP (9).
Diagnosis
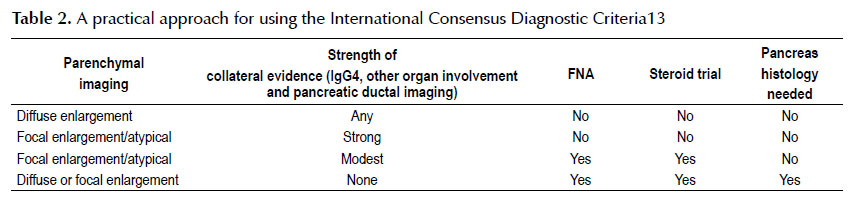
Different diagnostic criteria for AIP have been elaborated. The JapanPancreas Society criteria in 2002 (10), the Mayo clinic criteria in 2006 (11) and more recently the International Consensus Diagnostic Criteria (ICDC) in 2011 (12). The ICDC considers imaging, serology, other organ involvement, and response to steroids in order to provide a definitive or probable diagnosis (12). Table 2 shows a practical approach for using the ICDC (13).
Therapy
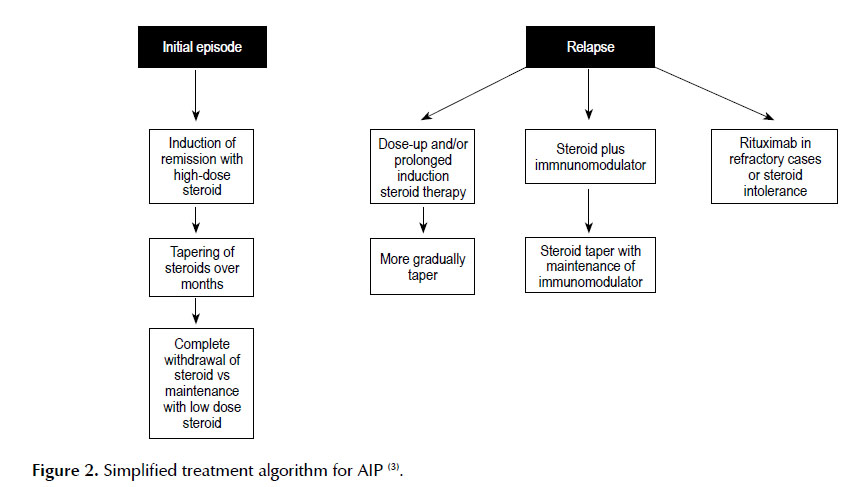
During an initial episode, induction of remission with high-dose steroids is typically usedi.e. prednisone 40 mg PO daily for four weeks. This is followed by a slow taper of the steroid over several months i.e. decreasing 5 mg per week. The Mayo clinic proposes complete withdrawal of the steroids while the Japanese society recommends to maintain patients with low-dose steroids in an attempt to prevent relapses. In case of relapse after finishing the course of steroid, the initial dose of prednisone be increased and/or the duration of the induction therapy can be prolonged, andthe taper can be done more gradually as well. Both the complete withdrawal of the steroid or maintenance with low dose steroid are reasonable options. Another therapeutic option in cases of relapse would be to start maintenance therapy with an immunomodulator such as azathioprine (2 mg/k/day), mercaptopurine (1 mg/k/ day) or mycophenolate mofetil (750 mg bid) along with prednisone. The steroid can be tapered off while maintaining the immunomodulatory (2,3). Hart et al did find a difference in relapse free survival between patients with relapsing AIP treated with prednisone alone vs. prednisone plus immunomodulatory (14). In refractory cases, rituximab, which is a monoclonal antibody directed against the CD 20 antigen on B-lymphocytes, can be used. In the same study, above performed at the Mayo clinic, found that 83% of the patients with relapsing AIP achieved remission with rituximab (10). See Figure 2 for a simplified therapeutic algorithm (3).
CONCLUSIONS
- AIP is uncommon but it should be in the differential diagnosis of any caseof pancreatitis.
- Physicians should be aware of the typical and atypical radiological features of AIP.
- Not every focal pancreatic mass on imaging is cancer.
- AIP can also present as a focal pancreatic mass.
- High IgG4 levels are suggestive but not diagnostic of type 1. High IgG4 levels can be seen in many other conditions.
- In not clear-cut cases, endoscopic ultrasound with FNA and/or trucut biopsy is recommended for an accurate diagnosis.
- AIP has an excellent response to steroids; nevertheless, relapses are common, and may need long term maintenance therapy.
REFERENCES
1. Okazaki K, Uchida K. Autoimmune pancreatitis: the past, present, and future. Pancreas. 2015;44(7):1006-16. [ Links ]
2. Hart PA, Zen Y, Chari ST. Recent advances in autoimmune pancreatitis. Gastroenterology. 2015;149(1):39-51. [ Links ]
3. Kamisawa T, Chari ST, Lerch MM, Kim MH, Gress TM, Shimosegawa T. Recent advances in autoimmune pancreatitis: type 1 and type 2. Gut. 2013;62(9):1373-80. [ Links ]
4. Manfredi R, Graziani R, Cicero C, Frulloni L, Carbognin G, Mantovani W, et al. Autoimmune pancreatitis: CT patterns and their changes after steroid treatment. Radiology.2008;247(2):235-43. [ Links ]
5. van Heerde MJ, Biermann K, Zondervan PE, Kazemier G, van Eijck CH, Pek C, et al. Prevalence of autoimmune pancreatitis and other benign disorders in pancreatoduodenectomy for presumed malignancy of the pancreatic head. Dig Dis Sci.2012;57(9):2458-65. [ Links ]
6. Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344(10):732-8. [ Links ]
7. van Heerde MJ, Buijs J, Hansen BE, de Waart M, van Eijck CH, Kazemier G, et al. Serum level of Ca 19-9 increases ability of IgG4 test to distinguish patients with autoimmune pancreatitis from those with pancreatic carcinoma. Dig Dis Sci 2014;59(6):1322-9. [ Links ]
8. Fujii LL, Levy MJ. EUS in the diagnosis of autoimmune pancreatitis. Pancreapedia: Exocrine Pancreas Knowledge Base [Internet]. 2013 [cited 2016 April 12]. doi: 10.3998/panc.2013.173. Available from: http://www.pancreapedia.org/?q=node/7391 [ Links ]
9. Chari ST, Kloeppel G, Zhang L, Notohara K, Lerch MM, Shimosegawa T, et al. Histopathologic and clinical subtypes of autoimmune pancreatitis: the Honolulu consensusdocument. Pancreas. 2010;39(5):549-54. [ Links ]
10. Okazaki K, Chiba T. Autoimmune related pancreatitis. Gut. 2002;51(1):1-4. [ Links ]
11. Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, Zhang L, et al. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol. 2006;4(8):1010-6. [ Links ]
12. Shimosegawa T, Chari ST, Frulloni L, Kamisawa T, Kawa S, Mino-Kenudson M, et al. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas. 2011;40(3):352-8. [ Links ]
13. Hart PA, Chari ST. Diagnosis Autoimmune pancreatitis. Pancreapedia: Exocrine Pancreas Knowledge Base [Internet]. 2013 [cited 2016 April 12]. doi: 10.3998/panc.2013.14. Available from: http://www.pancreapedia.org/?q=node/7328 [ Links ]
14. Hart PA, Topazian MD, Witzig TE, Clain JE, Gleeson FC, Klebig RR, et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience. Gut. 2013;62(11):1607-15. [ Links ]
Correspondence:
Jean A. Donet, MD,
Division of Gastroenterology, University of Miami. USA
E-mail: jean.donetmostacero@jhsmiami.org
Recibido: 08-02-2016
Aprobado: 29-04-2016
