










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Gastroenterología del Perú
versión impresa ISSN 1022-5129
Rev. gastroenterol. Perú vol.37 no.4 Lima oct./dic. 2017
REPORTE DE CASO
Feocromocitoma quístico gigante: reporte de un caso
Pheochromocytoma giant cystic: a case report
Rebeca Joya Vázquez1, Cristina Vecino Bueno1, José María Bengochea Cantos1, Olga Gómez García1, María Ángeles López López1, Antonio Molina Sánchez1, José Miguel Ruiz-Ayucar Imbert2, Elizabeth Barrera Melgarejo3
1 Servicio de Cirugía General, Hospital Campo Arañuelo. Navalmoral de la Mata, España.
2 Servicio de Anatomía Patológica, Hospital Campo Arañuelo. Navalmoral de la Mata, España.
3 Servicio de Cirugía General, Clínica San Felipe. Lima, Perú.
RESUMEN
El feocromocitoma quístico gigante es tumor adrenal raro en el que predomina el curso asintomático; por lo que muchos de los casos no son diagnosticados hasta el momento de la cirugía. La simple movilización del tumor se asocia con el paso a la sangre de grandes cantidades de catecolaminas y a una elevada morbimortalidad.; por esta razón la cirugía per se y su manejo perioperatorio constituyen un enorme desafío. En este artículo se presenta el caso de un feocromocitoma gigante maligno (35 cm) que ocupaba todo el hemiabdomen derecho. Aun con el diagnóstico preoperatorio de feocromocitoma, el bloqueo farmacológico preoperatorio y las medidas intraoperatorias, el paciente falleció poco antes de que finalizara la cirugía.
Palabras clave: Catecolaminas; Médula suprarrenal; Feocromocitoma (fuente: DeCS BIREME).
ABSTRACT
The giant cystic pheochromocytoma is a rare adrenal tumor in the predominantly asymptomatic course; so many cases are not diagnosed until the time of surgery. The simple mobilization of the tumor is associated with the passage to the blood of large amounts of catecholamines and high morbidity and mortality. So the surgery itself and perioperative management are a huge challenge. This article describes the case of a malignant giant pheochromocytoma (35 cm) which occupied the entire right abdomen. Even with the preoperative diagnosis of pheochromocytoma, pharmacological blockade preoperative and intraoperative measures, the patient died shortly before the end of surgery.
Keywords: Catecholamines; Adrenal medulla; Pheochromocytoma (source: MeSH NLM).
INTRODUCCIÓN
El feocromocitoma es un tumor suprarrenal usualmente benigno, que excepcionalmente excede los 2000 g de peso. Solo un 10% son extraadrenales; suele ser unilateral y no tiene predominancia de sexo. Su incidencia es de 1-2 por cada 100 000 habitantes por año, con un pico de máxima incidencia entre la 3ª y 4ª década de la vida. Su prevalencia es de 0,1% a 0,6% en pacientes hipertensos (1-5).
Los tumores de la médula adrenal secretan predominantemente norepinefrina, pese a que en la médula normal se produce el 80% de la epinefrina. Su presentación clínica depende de la secreción de catecolaminas. La hipertensión arterial sostenida o de curso paroxístico constituye el hallazgo más frecuente en estas neoplasias y la tríada sintomática clásica presente en más del 50% de los pacientes incluye: cefalea, palpitaciones y diaforesis (6). En muchos de los casos el paciente puede presentar un infarto al miocardio, insuficiencia cardíaca, edema agudo de pulmón, arritmias o un accidente cerebrovascular hemorrágico.
Los feocromocitomas gigantes (≈10%) frecuentemente presentan una degeneración quística y pueden desplazar órganos y estructuras vecinas; son clínicamente silentes por lo que su diagnóstico es incidental durante el acto quirúrgico, raramente exceden los 11-12 cm (7-9). El diagnóstico en ausencia del cuadro clínico típico suele ser difícil, aunque de vital importancia a fin de minimizar las complicaciones y reducir la mortalidad. La manipulación del tumor durante la cirugía se asocia a la liberación de una gran cantidad de catecolaminas (10). En este artículo se presenta el caso de un feocromocitoma gigante maligno que ocupaba todo el hemiabdomen derecho.
CASO CLÍNICO
En febrero 2016 es estudiado por el Servicio de Medicina Interna un paciente masculino de 75 años de edad con cuadro caracterizado por sensación de plenitud gástrica, inapetencia y pérdida de 10 kg de peso en los 3 meses previos. Sus únicos antecedentes de interés eran: ex-fumador, brucelosis, talasemia menor y adenoma prostático. En el examen físico se evidenció la presencia de una masa de consistencia blanda, no dolorosa de aproximadamente 30 cm que ocupaba todo el hemiabdomen derecho. Los estudios analíticos básicos fueron normales, excepto una discreta anemia: hemoglobina (10,3 g/dL), hematocrito (33,6%), un volumen corpuscular bajo (75) y una leve disminución de la actividad de pro-trombina (65%).
El TAC toracoabdominal evidenció una masa gigante (20 x 21 x 31 cm), en hipocondrio/flanco derecho con necrosis extensa, con un gran efecto de masa que infiltraba o dependía del polo superior del riñón derecho. También se observaron dos adenopatías retroperitoneales de 13 y 16 mm, respectivamente planteándose como primera opción diagnóstica un carcinoma suprarrenal (Figura 1). Los hallazgos de la resonancia fueron similares (Figura 2).
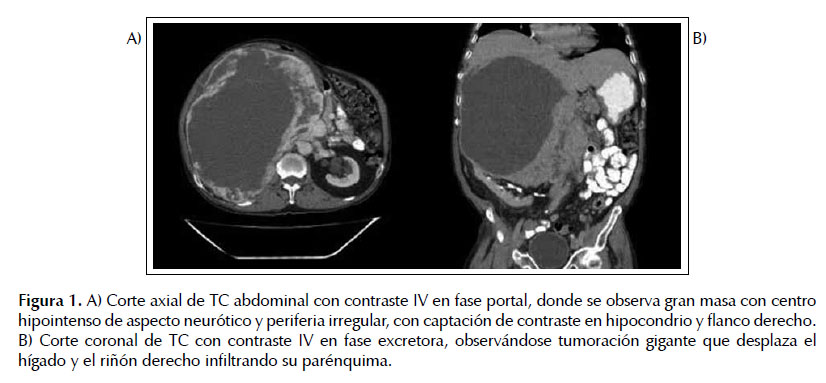
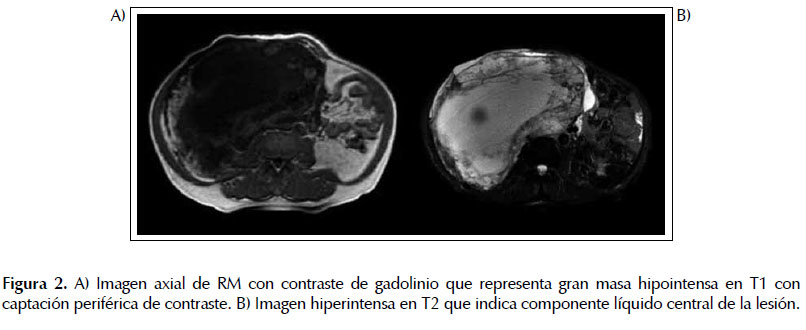
Ante la sospecha clínica de un feocromocitoma asintomático se realizaron determinaciones que permitieron confirmarlo: metanefrina: 2013 ug/24h, normetanefrina: 66 028 ug/24h, norepinefrina: 1384 pg/mL, y cortisol de 9,1 ug/dL. El paciente es valorado por el Servicio de Cirugía General de Aparato Digestivo planificándose cirugía de forma programada preferente y por el Servicio de Anestesiología indicándose bloqueo farmacológico preoperatorio primero Alfa adrenérgico y posteriormente Beta adrenérgico.
En marzo del 2016 ingresó electivamente para la cirugía. Bajo anestesia general, previa profilaxis antibiótica, antitrombótica y premedicación por parte del servicio de anestesiología, se realiza laparotomía subcostal derecha ampliada. Encontrándose como hallazgo un tumor retroperitoneal gigante de aproximadamente 35 x 24 x 20 cm, firmemente adherido a los segmentos VI y VII del hígado, que dependía de la suprarrenal derecha, generando un gran efecto de masa sobre el riñón ipsilateral y rodeando sin infiltrar la vena cava inferior en toda su circunferencia, en un tramo de 10 cm de longitud. La cirugía fue técnicamente compleja y muy laboriosa. Se liberaron las adherencias hepáticas, diseccionando el plano aortacava, uréter derecho y los vasos gonadales. Se diseca la vena cava, ligándose los pedículos renal y suprarrenal izquierdos. Debido al gran volumen de la masa, a su naturaleza quística y su disposición anatómica, fue necesario realizar punción-aspiración del tumor a fin de reducir su tamaño y así facilitar la disección, lo que coincide con descargas adrenérgicas, una elevación mantenida de la tensión arterial, inestabilidad hemodinámica y la imposibilidad de conseguir la hemostasia adecuada; en total, se drenaron aproximadamente 5 litros de un fluido cetrino. Pese al tratamiento indicado y a todas las medidas adoptadas fue imposible estabilizar el paciente quien fallece pocos minutos antes de que se terminara la cirugía; habiéndose completado casi en su totalidad la extirpación del tumor (Figura 3A).
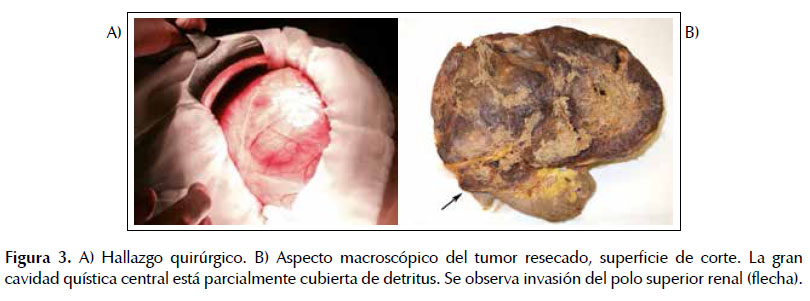
Anatomía patológica:
Tumor suprarrenal de 30 x 24 x 9 cm con superficie externa abollonada. Presentaba una gran cavidad quística central parcialmente colapsada, con un contenido serohemático. El componente sólido pesó 2170 g. La pared en torno al quiste medía 4 cm de espesor máximo, era de consistencia blanda y mos-traba una coloración gris rojiza, con zonas de edema y hemorragia (Figura 3B). El tumor estaba firmemente unido al riñón y reemplazaba gran parte de su polo superior. No se identificaron restos de la glándula su-prarrenal.
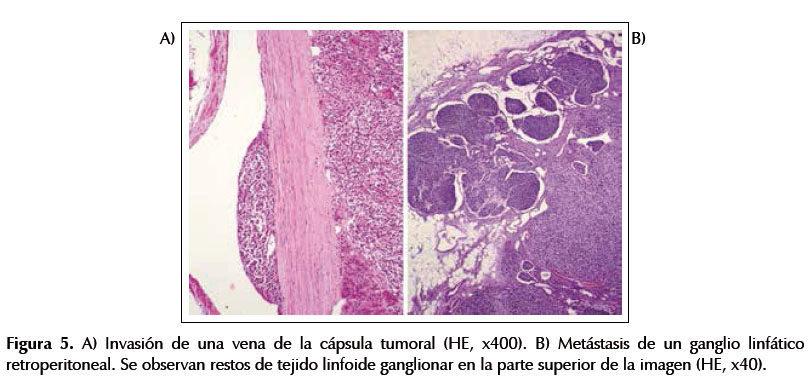
El estudio histológico confirmó el diagnóstico de feocromocitoma. La neoplasia formaba nidos y trabéculas separados por delgados tractos fibrovasculares y estaba constituida por células poligonales, con citoplasma eosinófilo o anfófilo, finamente granular. Se contaron 2 mitosis en 10 campos de gran aumento (Figura 4A). En el estudio inmunohistoquímico, las células tumorales expresaron marcadores neuroendocrinos: sinaptofisina, cromogranina y CD56 (Figura 4B). En la periferia del tumor eran visibles pequeños restos de corteza suprarrenal. Aunque la neoplasia estaba rodeada en su mayor parte por una cápsula fibrosa, invadía extensamente el polo superior renal, el tejido adiposo peritumoral y los vasos sanguíneos capsulares (Figura 5A). Se observó una metástasis en una adenopatía retroperitoneal de 1 x 1,3 cm, incluida en la pieza (Figura 5B). El diagnóstico final fue de feocromocitoma maligno.
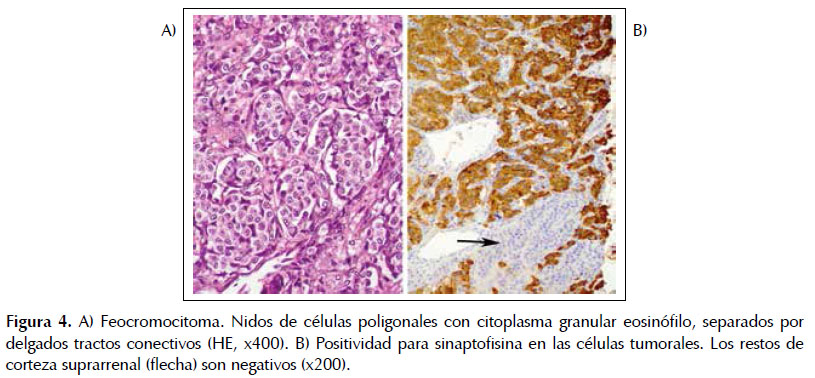
DISCUSIÓN
Las tumoraciones adrenales quísticas son muy infrecuentes. Su incidencia en series de autopsias es de 0,064-0,18%; y constituyen tan solo un 4-22% de la totalidad de los incidentalomas (11). El feocromocitoma excepcionalmente tiene un comportamiento maligno, y su variante quística suele ser aún más rara.
No suelen tener las características clínicas, radiológicas y analíticas típicas, pero si acompañarse de síntomas abdominales inespecíficos, pudiendo ser confundidos con otras neoplasias quísticas (12). Hasta la fecha pocos casos han sido publicados en la literatura. En una revisión hecha en el 2008 de 16 casos de feocromocitoma quístico, 6 pacientes tenían el cuadro clínico típico y otros 6, solo inestabilidad hemodinámica durante el acto quístico (13). Otra revisión de 15 casos hecha en el mismo año, reveló que la mayoría de ellos se presentaron en mujeres, la mitad de ellos sin síntomas, y la otra mitad tenían una analítica normal. Muchos de estos casos han sido erróneamente diagnosticados como tumores hepáticos o pancreáticos (13,14).
Se ha postulado que cuando el gran crecimiento tumoral no recibe el aporte sanguíneo requerido, tiene lugar una necrosis seguida de hemorragia. Finalmente, cuando el contenido se licua y es reabsorbido, el feocromocitoma adquiere la forma quística. Los pacientes con feocromocitoma quístico a menudo son asintomáticos y tanto la analítica del suero como de la orina es normal, porque las catecolaminas secretadas se metabolizan dentro de la neoplasia y sólo ocasionalmente una pequeña cantidad se libera en la circulación, no alcanzando niveles séricos ni urinarios elevados, pero si asociándose a la presencia de hipertensión arterial.
Los feocromocitomas varían en tamaño y peso. La media de peso de estos tumores es de 90 gr, según lo reportado por Sherwin, en una revisión de 96 casos (15). Es raro que los feocromocitomas gigantes pesen más de 1000 mg. En tres de los casos según Blessing et al., los tumores pesaron entre 2120 y 2500 g. Algunas publicaciones aisladas han reportado tumores de 1100-5900 gramos con un diámetro máximo de 12-22 cm (14). En nuestro caso fue necesario drenar durante la cirugía mediante la técnica de punción-aspiración cerca de 5000 cc del líquido del quiste, a fin de reducir el tamaño y facilitar la resección, reduciéndose su peso en seco a 2170 g para un peso total estimado de 7170 g; sus medidas (35 x 24 x 9 cm) también son superiores a la de los casos publicados. En nuestro caso además de ejercer un efecto de masa, la tumoración infiltraba el hígado, hecho también bastante infrecuente (14).
El diagnóstico por imagen puede hacerse mediante una tomografía computarizada con contraste, en el que la lesión muestra un marcado realce periférico y áreas de baja atenuación, en el intervalo de 5-15 unidades Hounsfield. En la resonancia magnética, las imágenes son más nítidas; las zonas de hemorragia aparecen hipointensas en T1 y tanto las zonas de necrosis y de hemorragia, hiperintensas en T2. En nuestro caso realizamos ambos estudios, pese a que la tomografía inicial orientó claramente a una neoplasia gigante dependiente de la suprarrenal derecha, planteando el diagnóstico de un feocromocitoma quístico silente, era necesario aclarar su relación con estructuras vasculares vecinas. El diagnóstico diferencial entre un quiste suprarrenal benigno y un feocromocitoma puede hacerse con una gammagrafía metaiodobenzilgua-nidina (MIBG), estudio que no se consideró necesario en este caso. La certeza diagnóstica solo la confiere la histología y especialmente la inmunohistoquímica.
La liberación de catecolaminas en cualquier momento de la evolución y en especial durante la manipulación del tumor en la cirugía de resección, puede ocasionar shock, fiebre, leucocitosis y/o una crisis hipertensiva de difícil manejo (13). En el presente caso la manipulación del tumor generó un cuadro de inestabilidad hemodinámica incontrolable. Por lo que el paciente falleció poco antes de finalizar la cirugía.
En conclusión, el feocromocitoma quístico gigante es una entidad muy poco frecuente existiendo pocos reportes de casos mayores de 20 cm. A pesar de la sintomatología inespecífica de la enfermedad, la especificidad de los métodos de estudio hace que el diagnóstico de feocromocitoma casi siempre se realice de forma preoperatoria, por lo que es esencial incluirlo en el diagnóstico diferencial de cualquier tumoración suprarrenal quística de gran tamaño.
Este caso es un ejemplo del desafío que constituye la resección de feocromocitomas quísticos de grandes dimensiones.
Conflictos de intereses: Los autores declaran no tener ningún conflicto de interés.
Fuente de financiamiento: Ninguna.
REFRENCIAS BIBLIOGRÁFICAS
1. Recasens M, Oriola J, Fernández-Real JM, Roig J, Rodríguez-Hermosa JI, Font JA, et al. Asymptomatic bilateral adrenal pheochromocytoma in a patient with a germline V804M mutation in the RET proto-oncogene. Clin Endocrinol (Oxf). 2007;67(1):29-33. [ Links ]
2. Lavin N. Endocrinología y metabolismo. Madrid: ed. Marban Libros S.L; 2003. [ Links ]
3. Eisenhofer G, Walther MM, Huynh TT, Li ST, Bornstein SR, Vortmeyer A, et al. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab. 2001;86(5):1999-2008. [ Links ]
4. Dluhy R, Lawrence J, Williams RH. Hipertensión de origen endocrino. En: RH Williams. Tratado de En-docrinología Clínica. 10ª ed. USA: Ed. Elservier; 2000. p. 677. [ Links ]
5. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Pheochromocytoma. Lancet. 2005;366(9486):665-75. [ Links ]
6. Cotesta D, Petramala L, Sierra V, Pergolini M, Crescenzi E, Zinnamosca L, et al. Clinical experience with pheocromocytoma in a single centre over 16 years. High Blood Press Cardiovasc Prev. 2009;16(4):183-93. [ Links ]
7. Saveswaran V, Kumar S, Kumar A, Vamseedharan M. A giant cystic pheochromocytoma mimicking liver abscess an unusual presentation –a case report. Clin Case Resp. 2015;3(1):64-8. [ Links ]
8. Galvao-Braga C, Riveiros S, Martins J, Arantes C, Ramos V, Primo J, et al. Pheochromocytoma diag-nosed after anticoagulation for atrial fibrillation ablation procedure: a giantin disguise. Rev Port Cardiol. 2014;33(4):245. [ Links ]
9. Wang HL, Sun BZ, Xu ZJ, Lei WF, Wang XS. Undiagnosed giant cystic pheochromocytoma: a case report. Oncol Left. 2015;10(3):1444-6. [ Links ]
10. Melegh Z, Rényi-Vamos F, Tanyay Z, Koves I, Orosz Z. Giant cystic pheochomocytoma located in the renal hilus. Pathol Res Pract. 2002;198:103-6. [ Links ]
11. Bellantone R, Ferrante A, Raffaelli M, Boscherini M, Lombardi CP, Crucitti F. Adrenal cystic lesions: report of 12 surgically trated cases and review of the literature. J Endocrinol Invest. 1998;21(2):109-14. [ Links ]
12. Grozinsky-Glasberg S, Szalat A, Benbassat CA, Gorshtein A, Weinstein R, Hirsch D, et al. Clinically silent chromaffin-cell tumors: tumor characteristics and long term prognosis in patients with incidentally dis-covered pheochromocytomas. J Endocrinol Invest. 2010;33(10):739-44. [ Links ]
13. Antedomenico E, Wascher RA. A case of mistaken identity: giant cystic pheochromocytoma. Curr Surg. 2005;62(2):193-8. [ Links ]
14. Wu JS, Ahya SN, Reploeg MD, Singer GG, Brennan DC, Howard TK, et al. Pheocromocytoma presenting as a giant cystic tumor of the liver. Surgery. 2000;128(3):482-4. [ Links ]
15. Sherwin RP. Histopathology of pheochromocytoma. Cancer. 1959;12(5):861-77. [ Links ]
Correspondencia:
Rebeca Joya
E-mail: rebecajoyav@gmail.com
Recibido: 12-11-2016
Aprobado: 13-4-2017

