










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Gastroenterología del Perú
versión impresa ISSN 1022-5129
Rev. gastroenterol. Perú vol.38 no.2 Lima abr./jun. 2018
REPORTE DE CASO
Malformación de Abernethy: causa inusual de cianosis central en pediatría
Abernethy malformation: unusual cause of central cyanosis in pediatrics
Juan Ampuero E.1,a, Guillermo Bernaola2,b, Julio Arbulú2,b, Erick Salas2,b
1 Neumología Pediátrica, Hospital Edgardo Rebagliati Martins. Lima, Perú.
2 Unidad de Neumología Pediátrica, Hospital Edgardo Rebagliati Martins. Lima, Perú.
a Residente, b Asistente
RESUMEN
El shunt porto sistémico congénito es una causa rara de hipoxemia y una patología muy poco frecuente con complicaciones severas si no es tratada. Fue descrito por primera vez por John Abernethy en 1793. Existen dos tipos: tipo I (shunt termino lateral) en el que hay ausencia total del flujo portal intrahepático y tipo II (shunt latero lateral) con flujo portal parcialmente conservado. Se presenta el caso de una niña de 6 años de edad con antecedente de hipoxemia crónica desde los 4 años y medio de vida, acompañado de disnea progresiva, quien fue referida a la unidad de neumología pediátrica con diagnóstico de cianosis central. Entre los estudios diagnósticos considerados se realizó ecografía doppler del sistema venoso portal, evidenciándose ausencia de vena porta principal; además se realizó angiotomografía del sistema arterio-venoso portal y mesentérico, confirmándose la agenesia de vena porta. Se completó el estudio con una porto-esplenografía que confirmó el diagnóstico de malformación de Abernethy tipo I b. La malformación de Abernethy tipo I es más frecuente en el sexo femenino, tiene varias formas de presentación y el tratamiento es el trasplante hepático. En la malformación de Abernethy tipo II la circulación portal es variable y tiene mejor pronóstico que la de tipo I. La disnea al ejercicio y la cianosis central es una forma de presentación que debemos tener en cuenta en el diagnóstico diferencial de la patología cardiorrespiratoria en la edad pediátrica.
Palabras clave: Agenesia; Vena porta; Niño (fuente: DeCS BIREME).
ABSTRACT
The congenital portosystemic shunt is an uncommon disease with severe complications if not treated. This rare cause of hypoxemia was first described by John Abernethy in 1973. There are two types: type I (termino-lateral shunt), in which there is total absence of the intrahepatic portal flow, and type II (latero-lateral shunt), in which the portal flow is partially preserved. We present the case of a 6-year-old girl with chronic hypoxemia history since 4 and a half years of age, showing progressive dyspnea, who was referred to the Pediatric Pulmonary Division with the diagnosis of central cyanosis. An Echo-Doppler in the portal venous system was performed, reporting agenesis of the principal portal vein. This finding was corroborated by an angiography of the portal and mesenteric arteriovenous system. The study was completed with a portosplenography, which confirmed the diagnosis of type Ib Abernethy malformation. The type I Abernethy malformation is more common in females, shows up in different ways and is treated with liver transplantation. On the other hand, type II Abernethy malformation shows a variable portal circulation and has a better prognosis than type I. Dyspnea when exercising and central cyanosis should be considered to make a differential diagnosis of cardiorespiratory disease at a pediatric age.
Keywords: Agenesis; Portal vein; Child (source: MeSH NLM).
INTRODUCCIÓN
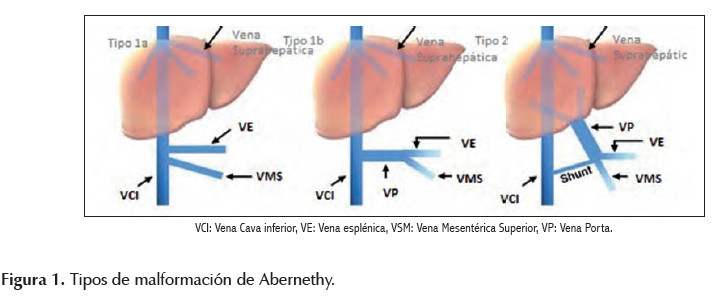
El shunt porto sistémico congénito o malformación de Abernethy es una entidad sumamente rara, en la cual el flujo sanguíneo porto sistémico drena en una vena sistémica, evitando así su paso por el hígado través de un shunt parcial o completo. Así se diferencia dos tipos: el tipo I (shunt término lateral) en el que hay ausencia total de flujo portal intrahepático y el tipo II (shunt latero lateral) con flujo portal parcialmente conservado (Figura 1). La malformación tipo I es la más frecuente, predomina en el sexo femenino y suele asociarse a otras malformaciones como poliesplenia y cardiopatía, siendo la trisomía 21 la alteración cromosómica a la que se asocia con mayor frecuencia (1,2,3,4). Hay varias formas de presentación y la mayoría de niños pueden estar inicialmente asintomáticos; sin embargo, a largo plazo pueden aparecer nódulos hepáticos por aumento del flujo arterial y disminución del flujo portal y alteraciones hemodinámicas por hiperflujo (3,5,6,7). Se postula que la cianosis es secundaria a un desequilibrio del cociente ventilación perfusión causado por hiperflujo pulmonar (8). La diferenciación en tipo I ó tipo II es esencial para determinar el tratamiento definitivo, el cual será trasplante hepático en la malformación tipo I. El objetivo de este reporte es resaltar y considerar a la malformación de Abernethy como causa inusual de disnea y cianosis central en la población pediátrica.
CASO CLÍNICO
Paciente de sexo femenino de 6 años de edad, con tiempo de enfermedad de un año y medio que inicia de forma insidiosa con disnea y cianosis. La disnea es progresiva, inicialmente es a grandes esfuerzos para finalmente ser en reposo. La cianosis también fue progresiva. Acude en tres oportunidades a un hospital de su localidad ingresando por emergencia donde le indican oxígeno y nebulizaciones con salbutamol con poca mejoría. Por ser los síntomas más frecuentes es evaluado por médico cardiólogo quien nota disnea en reposo y cianosis marcada en labios y lecho ungueal, además nota retraso en la curva de talla y peso para la edad, por lo cual decide referirla al INCOR (Instituto Nacional del Corazón) para descarte de cardiopatía congénita. En el INCOR realizan una ecocardiografía y cateterismo cardiaco, no evidenciándose malformaciones cardiacas ni hipertensión pulmonar, por lo cual es referida a la unidad de Neumología Pediátrica del Hospital Edgardo Rebagliati Martins para descarte de patología respiratoria. En la unidad de Neumología se elabora un plan de trabajo que incluye radiografía de tórax, dosaje de metahemoglobina, prueba de hiperoxia, ecocardiografía, ecografía abdominal, ecodoppler abdominal, angiotem abdominal y por último esplenoportografía.
Examen físico y evolución:
Paciente con cianosis central, saturación de oxigeno de 75% sin aporte de oxígeno y que llega hasta 83% con oxígeno a través cánula nasal a 3 L/min. Piel tibia con escaso panículo adiposo, algunas telangiectasias en cara e hipocratismo digital marcado. La relación T/E corresponde al p10 y P/E al p5; tórax simétrico, no se evidencia dificultad respiratoria, el murmullo vesicular pasa bien en ambos campos pulmonares sin ruidos agregados. A nivel cardiovascular no se auscultan soplos. En abdomen no hay visceromegalia, ni se observan signos de hipertensión portal.
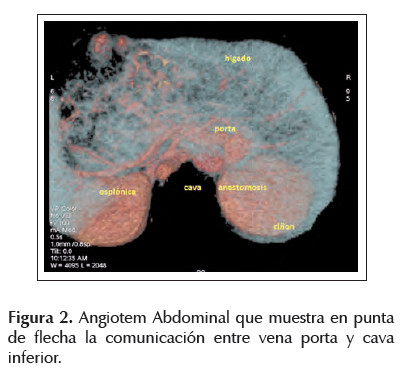
Al ingreso se dosó metahemoglobina cuyo resultado fue 0,5% (dentro del rango normal), la radiografía de tórax y la ecocardiografía fueron normales. El ecodoppler abdominal informó la existencia de shunt entre vena porta derecha y vena cava. Se solicitó angiotem abdominal con reconstrucción vascular confirmándose el hallazgo del shunt (Figura 2). Luego se realizó una porto esplenografía que demostró la presencia de flujo sanguíneo con comunicación entre vena porta y vena cava inferior e inexistencia de la vena porta izquierd. De esta manera todo el sistema venoso esplácnico no pasa por el hígado, dirigiendo toda la sangre a vena cava inferior (Figura 3).
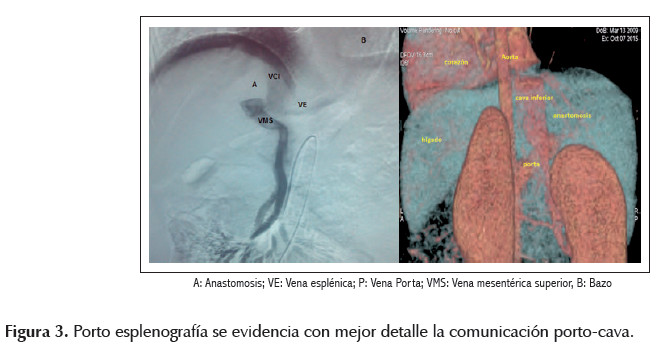
El diagnóstico final fue malformación de Abernethy tipo Ib y se refirió al paciente al servicio de cirugía pediátrica para trasplante hepático.
DISCUSIÓN
Las malformaciones del sistema venoso abdominal son alteraciones vasculares raras. Esta anomalía vascular varía mucho en su presentación y el tratamiento dependerá del tipo de malformación (1,2,7,9). El primer hallazgo descrito por Abernethy, ausencia congénita de la vena porta y shunt mesentérico-cava, se basó en un examen post-morten realizado a un lactante de 10 meses de edad quien murió por causa desconocida (4). En 1883, kiernan describió el segundo caso de anastomosis porto cava congénita en una adolescente de 18 años de edad en quien la arteria hepática se observó elongada (10).
Se ha publicado en la literatura mundial 39 casos de Abernethy tipo I y 22 casos de Abernethy tipo II (13). En el tipo I hay una ausencia total del flujo portal intrahepático, estos pacientes llegan a encefalopatía hepática en edad adulta, y el tratamiento es el trasplante hepático.
Un reporte caso publicado el 2005 que incluyo 4 pacientes, de los cuales tres eran varones y 1 mujer, fueron diagnosticados entre los 0 y 28 meses de edad con una mediana de 14 meses, tres de los casos correpondieron al shunt porto sistémico tipo I y uno (varón) al tipo II. En todos estos casos el diagnóstico se hizo mediante ecodoppler abdominal. Sólo un paciente varón de 2 años presentó cianosis como hallazgo, siendo el debut de este paciente un cuadro de colecistitis asociado a colelitiasis, hipercolesterolemia e hipercalciurica. En el resto de pacientes el modo de presentación fue diferente (1).
En un reporte de una serie de casos de veintidós niños tratados y seguidos en el hospital de Bicetre con derivación portosistémica, donde se evaluó la evolución y su comportamiento; este estudio incluyó la medición de la presión de la Porta, angiografía Portal durante la prueba de oclusión y el cierre de la derivación por vía quirúrgica y/o técnicas endovasculares. Los resultados fueron variados, cinco de los recién nacidos con shunt intrahepático presentó colestasis que resolvió espontáneamente, 17 niños mayores presentaron tumoraciones en el hígado y tuvieron signos de hipertensión portal, hipertensión arterial pulmonar, encefalopatía porto sistémica, insuficiencia cardíaca y glomerulonefritis. Las derivaciones porto sistémicas fueron cerradas por métodos endovasculares en 5 niños y quirúrgicamente en 10 niños. El cierre del Shunt dio lugar a la restauración del flujo portal intrahepático en todo su sistema con regresión completa o parcial de masas hepáticas benignas además de regresión y estabilización pulmonar, cardíaca, neurológica y renales; cabe mencionar que tan solo tres niños de entre los 5 y 11 años de edad, presentaron hipertensión pulmonar, incluyendo disnea de esfuerzo. Además dos niños presentaron hipoxemia secundaria a la derivación arterio venosa pulmonar estos pacientes tenían cianosis y disnea con saturación de oxigeno que oscilaba entre 83 y 90% (11).
Otra forma de presentación, menos frecuente, es con cianosis central, como es el caso de 2 pacientes de la India, de 5 y 9 años de edad qienes acudieron a atención medica por cianosis como signo principal además de retardo de crecimiento. El diagnostico de ambos pacientes se realizó en gase a una ecografía dopplern que mostró la anastomosis de la vena Porta con la vena Cava Inferior (4,8).
CONCLUSIÓN
El shunt porto sistémico congénito es una malformación rara que tiene múltiples formas de presentación y se asocia a otras malformaciones. La disnea y la cianosis central es una forma de presentación poco usual que debemos tener en cuenta en el diagnóstico diferencial de patología cardiorrespiratoria en pediatría.
La derivación porto sistémica congénita conlleva riesgos de complicaciones graves en los niños. Se debe considerar de forma preventiva el cierre de una derivación que persiste más alla de los de 2 años de edad.
Es necesario el diagnóstico precoz. El examen preferente es el ecodoppler con posterior confirmación medianfe angiotem abdominal. El tratamiento es sumamente importante pues su retraso puede devenir en lesiones irreparables hasta la insuficiencia hepática y muerte.
REFERENCIAS BIBLIOGRÁFICAS
1. Ávila LF, Luis AL, Encinas JL, Hernández F, Olivares P, Fernández Cuadrado J, et al. Shunt porto cava congénito. Malformación de Abernethy. Cir Pediatr. 2006;19:204-9. [ Links ]
2. Nso AP, Garcia P, Quero J. Malformaciones vasculares abdominales y Síndrome de Down. An Pediatr (Barc). 2007;66(4):410-2. [ Links ]
3. Saxena AK, Sodhi KS, Arora J, Thapa BR, Suri S. Congenital intrahepatic portosystemic venous shunt in an infant with Down syndrome. AJR Am J Roentgenol. 2004;183(6):1783-4. [ Links ]
4. Abernethy J. Account of two instances of uncommon formation in the viscera of the human body. Philos Trans R Soc. 1793;83:59-66. [ Links ]
5. Nso AP, Garcia P, Quero J. Malformaciones vasculares abdominales y Síndrome de Down. An Pediatr (Barc). 2007;66(4):410-2. [ Links ]
6. Courtens W, Segers V, Johansson A, Avni FE. Association between Down syndrome and portohepatic shunt. Am J Med Genet. 2000;93(2):166-8. [ Links ]
7. Witters P, Maleux G, George C, Delcroix M, Hoffman I, Gewillig M, et al. Congenital veno-venous malformations of the liver: widely variable clinical presentations. J Gastroenterol Hepatol. 2008;23(8 Pt 2):e390-4. [ Links ]
8. Jagadisan B, Krishnamurthy S, Raghavan R, Barathi SD. Chronic hypoxemia in a child: thinking outside the box. Indian Pediatr. 2014;51(10):829-30 [ Links ]
9. Morgan G, Superina R. Congenital absence of the portal vein: two cases and a proposed classification system for portasystemic vascular anomalies. J Pediatr Surg. 1994;29(9):1239-41. [ Links ]
10. Kierman F. The anatomy and physiology of the liver. Philos Trans 1833;113:711-70. [ Links ]
11. Franchi-Abella S, Branchereau S, Lambert V, Fabre M, Steimberg C, Losay J, et al. Complications of congenital portosystemic shunts in children: therapeutic options and outcomes. J Pediatr Gastroenterol Nutr. 2010;51(3):322-30. [ Links ]
12. Sahu MK, Bisoi AK, Chander NC, Agarwala S, Chauhan S. Abernethy syndrome, a rare cause of hypoxemia: a case report. Ann Pediatr Cardiol. 2015;8(1):64-6. [ Links ]
13. Murray CP, Yoo SJ, Babyn PS. Congenital extrahepatic portosystemic shunts. Pediatr Radiol. 2003;33(9);614-20. [ Links ]
Correspondencia:
Juan Ampuero E.
E-mail: ampuero57@hotmail.com
Recibido: 07-08-2017
Aprobado: 11-12-2017

