










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Gastroenterología del Perú
versión impresa ISSN 1022-5129
Rev. gastroenterol. Perú vol.38 no.3 Lima jul./set. 2018
ARTÍCULO DE REVISIÓN
Síndrome de Lynch: aspectos genéticos, clínicos y diagnósticos
Lynch syndrome: genetic, clinical and diagnostic aspects
María del Carmen Castro-Mujica1,2,a, Claudia Barletta-Carrillo3,4,b,c
1 Docente de la Facultad de Medicina, Universidad Ricardo Palma. Lima, Perú.
2 Clínica Internacional. Lima, Perú.
3 Laboratorio de Genética Humana, Universidad Nacional Mayor de San Marcos. Lima, Perú.
4 Laboratorio de Biología Molecular, Laboratorio Clínico Roe. Lima, Perú.
a Médico genetista, b Bióloga, c Magíster en Biología Molecular
RESUMEN
Esta revisión tiene como objetivo dar a conocer los aspectos genéticos, clínicos y diagnósticos del síndrome de Lynch, además de brindar la información más relevante acerca de la asesoría genética en estos pacientes y las recomendaciones actuales para su seguimiento.
Palabras clave: Síndrome de Lynch; Neoplasias colorrectales hereditarias sin poliposis; Síndromes neoplásicos hereditarios (fuente: DeCS BIREME)
ABSTRACT
This review aims to present the genetic, clinical and diagnostic aspects of Lynch syndrome, as well as providing the most relevant information about genetic counseling in these patients and the current recommendations for their surveillance.
Keywords: Lynch syndrome; Colorectal neoplasms, hereditary nonpolyposis; Neoplastic syndromes, hereditary (source: MeSH NLM).
INTRODUCCIÓN
El Síndrome de Lynch (SL) (MIM #120435) (1), también llamado cáncer colorrectal (CCR) hereditario no polipósico (CCRHNP), es un síndrome genético, heterogéneo, con patrón de herencia autosómico dominante y penetrancia incompleta. Este síndrome predispone al desarrollo de CCR, principalmente, así como a neoplasias extracolónicas como el cáncer de endometrio, ovario, intestino delgado, estómago, uréter, vía biliar, páncreas, próstata, entre otros (2). El SL es causa del 1-3% del total de casos de CCR, 0,8-1,4% de casos de cáncer de endometrio y su prevalencia en la población se estima en 1:440 (3).
El SL se debe a variantes patogénicas germinales en alguno de los genes involucrados en la reparación de los errores de la replicación del ADN (MMR, del inglés mismatch repair) (MLH1, MSH2, MSH6, PMS2), así como a deleciones germinales del gen EPCAM, el cual no es un gen MMR pero causa la inactivación del gen MSH2 (2,4). Aproximadamente el 90% de casos de SL se deben a variantes patogénicas germinales en los genes MLH1 y MSH2, seguido de MSH6 en el 7%-10% de los casos y PMS2 en menos de 5% de los casos; mientras que las deleciones en el gen EPCAM son la causa en ~1% de los casos (3).
El diagnóstico clínico-genético de este síndrome requiere de la evaluación de la historia personal y familiar del paciente con CCR y/u otra neoplasia extracolónica asociada al SL, además del estudio molecular que identifique la alteración a nivel germinal en alguno de los genes relacionados al SL (5). El diagnóstico oportuno de SL permite brindar la asesoría genética al paciente y familiares en riesgo y establecer medidas de seguimiento de manera multidisciplinaria, así como el planteamiento de estrategias de prevención adecuadas a fin de evitar la morbimortalidad por cáncer.
Esta revisión tiene como objetivo dar a conocer los aspectos clínicos y métodos de diagnóstico del SL, además de brindar la información más relevante acerca de la asesoría genética en estos pacientes y las recomendaciones más relevantes para su seguimiento.
HISTORIA DEL SÍNDROME DE LYNCH
La historia del SL inicia en el año 1895, cuando el Dr. Aldred Warthin realiza un árbol genealógico de su costurera quien estaba convencida que fallecería a causa del cáncer, debido a que poseía el antecedente de múltiples familiares con cáncer (6). Esta familia fue denominada "Familia G" por la alta incidencia de cáncer gastrointestinal. La joven falleció debido a un cáncer de endometrio metastásico y estos hallazgos fueron publicados por el Dr. Warthin en el año 1913 (7).
En el año 1962, el Dr. Henry Lynch evaluó a un paciente en recuperación de delirium tremens quien refirió haber bebido alcohol en exceso por tener la certeza que iba a fallecer por cáncer de colon, el cual era muy prevalente en su familia (8). El Dr. Lynch elaboró el árbol genealógico de la familia del paciente, encontrando antecedentes de cáncer de colon, endometrio y ovario, y que además se presentaban con un patrón de herencia autosómico dominante (8). Los reportes de patología de los familiares afectados de cáncer de colon revelaron ser algunos sincrónicos, otros metacrónicos, y no mostraban evidencia de adenomas colónicos, hecho que descartaba la presencia de un síndrome de poliposis colónica familiar (8). Durante el Congreso de la Sociedad Americana de Genética Humana en el año 1964, el Dr. Lynch presentó en una comunicación oral los hallazgos de la historia familiar del paciente evaluado (8). Esta familia fue denominada "Familia N" por ser de Nebraska. A este congreso, asistió la Dra. Marjorie Shaw, médico genetista de la Universidad de Michigan, quien se mostró interesada en el reporte del Dr. Lynch, debido a que ella había evaluado a una familia similar, y deseaba hacer un estudio en colaboración con el Dr. Lynch (8). La familia evaluada por la Dra. Shaw fue denominada "Familia M" por ser de Michigan. En el año 1966, el Dr. Lynch y la Dra. Shaw junto a sus colaboradores publicaron los hallazgos de ambas familias (9).
El Dr. James French, médico sucesor del Dr. Aldred Warthin, tuvo conocimiento sobre la investigación del Dr. Lynch y la Dra. Shaw en cuanto a las familias N y M (10) y ofreció al Dr. Lynch el acceso a los datos recopilados por el Dr. Warthin, incluyendo los bloques y láminas correspondientes a los tejidos tumorales de los afectados en la Familia G, así como los bosquejos del árbol genealógico elaborado (8). El Dr. Lynch y una trabajadora social viajaron en múltiples oportunidades a Ann Arbor para revisar los registros del Dr. Warthin, y con la colaboración del patólogo Dr. Arthur Larsen revisaron nuevamente las láminas y reportes de patología de algunos de los casos afectados de cáncer (8). En el año 1971 se publicó una revisión acerca de los descendientes de la Familia G con los nuevos datos obtenidos (11).
Posteriormente, el Dr. Lynch y sus colaboradores identificaron más familias que presentaban características similares (8). Estos hallazgos motivaron al Dr. Lynch a desarrollar el Servicio de Información a la Familia (FIS, del inglés Family Information Service) (12) que consistía en la evaluación de familias del Oeste de EEUU mediante el uso de un vehículo personalizado el cual poseía una sala de entrevista, otra de examen físico y un pequeño laboratorio para extracción de sangre, permitiéndoles ir a las zonas donde residían todos los miembros de las familias evaluadas y educar a los familiares en riesgo y médicos de esas zonas, explicando el componente genético en la familia y la importancia del seguimiento de la familia. Este hecho marcó un hito en el diagnóstico y asesoría genética en los casos de cáncer hereditario, planteando la importancia de la evaluación integral del paciente.
En el año 1977, el Dr. Lynch realizó una publicación donde describía que los pacientes afectados presentaban el cáncer a una edad temprana, aproximadamente 20 a 25 años antes que los casos esporádicos, con un promedio de 44 años de edad para el desarrollo del CCR, además de ser preferentemente en colon proximal (13), presentarse de forma sincrónica o metacrónica, y un mayor riesgo a múltiples neoplasias primarias principalmente de CCR y endometrio (14).
En el año 1990 se constituyó un grupo cooperativo llamado InSIGHT (del inglés, International Society for Gastrointestinal Hereditary Tumours Incorporated), a fin de promover el estudio de este síndrome a nivel internacional (10). Inicialmente, el Dr. Lynch denominó a estos casos como "Síndrome de cáncer familiar" (8). Posteriormente, este término fue cambiado al de "CCR Hereditario No Polipósico" (CCRHNP), sin embargo como existía la posibilidad de desarrollarse neoplasias extracolónicas y/o algunos pólipos colónicos (15), se denominó "Síndrome de Lynch" (16), el cual se mantiene hasta la actualidad.
VÍA DE MOLECULAR DEL SISTEMA MMR
La inestabilidad microsatelital producto de la falla del sistema MMR es la característica principal de los tumores en pacientes con SL (17). El sistema de reparación MMR es un mecanismo de reparación indirecto debido a que involucra lesiones que ocurren durante la síntesis de nuevo ADN y toma lugar finalizado el proceso de síntesis, siendo esencialmente un proceso post-replicativo pero asistido por proteínas que forman parte de la maquinaria de replicación (18). Ver Tabla 1.
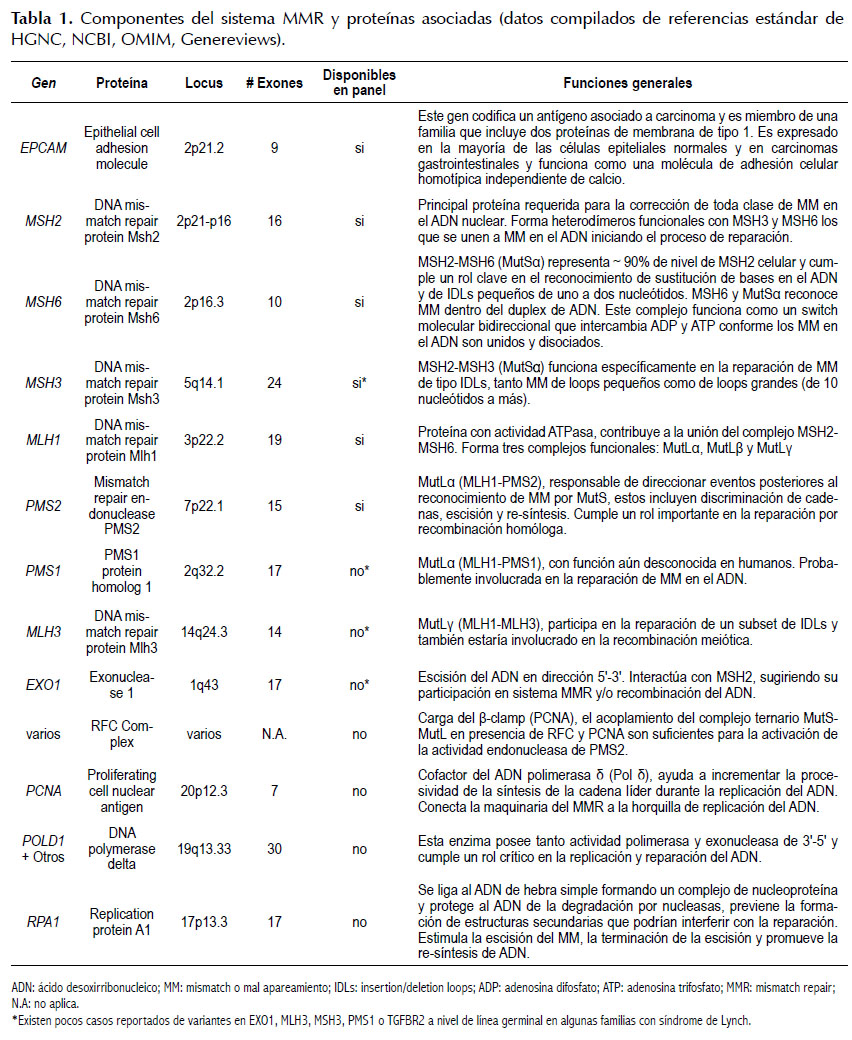
El sistema MMR reconoce y repara pequeños loops dentro de la doble hebra del ADN originados por la incorporación errónea de nucleótidos tanto por mal apareamiento base-base o por loops de inserción/ deleción (IDLs) (18). Cuando existen fallas en el sistema MMR debido a mutaciones germinales en alguno de los genes del MMR (MLH1, MSH2, MSH6 y PMS2), las células deficientes en MMR presentan un incremento en 100-1000 veces de la tasa de error durante la replicación, y a esto se le conoce usualmente como fenotipo mutador (19).
El sistema MMR es responsable de la vigilancia y corrección de errores introducidos dentro de las secuencias microsatélites por la ADN polimerasa. Los microsatélites son repeticiones cortas en tándem de una a seis bases de longitud que se encuentran dispersas a través de todo el genoma humano, estimándose cerca de medio millón de loci microsatélites (18). Dado que algunos genes incluyen secuencias microsatélites dentro de sus regiones codificantes, la IMS en ellos resulta en la producción de proteínas no funcionales o truncas afectándose importantes procesos celulares como la apoptosis, reparación del ADN, regulación transcripcional, translocación y modificaciones de proteínas entre otras. La IMS intragénica por ejemplo conduce a la inactivación del gen supresor de tumor TGF -RII en cerca del 80% de tumores MMR deficientes, mientras que el 20% restante presenta inactivo a IGFRII también debido al mismo mecanismo. Otro gen importante que aparece inactivo por IMS intragénica es el promotor de apoptosis BAX (18,19).
El sistema de MMR puede ser dividido en cuatro pasos (18-20): Ver Figura 1.
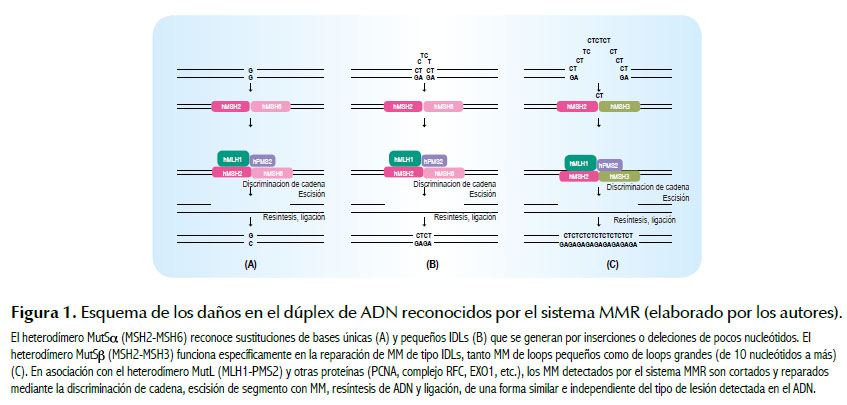
1) Reconocimiento del mismatch (MM) dentro del dúplex de ADN repetido por las proteínas MutS, las cuales se encuentran formando los heterodímeros MutS (MSH2-MHS6) y MutS (MSH2-MSH3) que reconocen lesiones específicas en el ADN, siendo MutS responsable de la reparación de mismatches base-base o pequeños IDLs y MutS funciona en la reparación tanto de IDLs pequeños y grandes (10 nucleótidos a más).
2) Reclutamiento de enzimas de reparación de las lesiones MM en el ADN, esto es la unión de MutL (MLH1-PMS2), involucrada en la reparación de una amplia variedad de MM en humanos. Este reclutamiento mejoraría notablemente la eficiencia de MutS en el reconocimiento de MM en el ADN dúplex.
3) Escisión de base MM o secuencia incorrecta, mediante la interacción de MutS y MutS con PCNA, este último con un rol importante dentro de la maquinaria del sistema MMR en la discriminación entre las dos cadenas del DNA dúplex. Adicionalmente PCNA también interactúa con la enzima Exo1, responsable de la excisión del MM y la resíntesis del ADN.
4) Resíntesis del ADN a partir de la cadena molde parental por la ADN polimerasa.
ASPECTOS CLÍNICOS DEL SÍNDROME DE LYNCH
Criterios de Amsterdam y Bethesda
El diagnóstico del SL resulta un reto debido a la gran heterogeneidad clínica que posee. Por esta razón, se han establecido distintos criterios clínicos que nos permiten sospecharlo. Ver Tabla 2. En el año 1991, el Grupo Colaborativo Internacional de Investigadores establecieron los Criterios de Amsterdam I (21) con la intención de asegurar que los investigadores a nivel mundial siguieran los mismos criterios en la clasificación de los pacientes a evaluar. Posteriormente, en el año 1998 se establecieron los criterios de Bethesda (22) para determinar a los candidatos al estudio de inestabilidad de microsatélites (IMS) y/o inmunohistoquímica (IHQ), siendo estos los estudios preliminares al secuenciamiento génico. En estos casos, sólo se requiere del cumplimiento de al menos uno de los criterios de Bethesda. Debido a que los Criterios de Amsterdam I eran muy estrictos y excluían a familias con pocos miembros o con manifestaciones extracolónicas, en el año 1999 se desarrollaron los criterios de Amsterdam II (23). Además, se estableció que para el cumplimiento de los criterios Amsterdam II se requería poseer los cuatro criterios establecidos (5). Finalmente en el año 2004, se propusieron los criterios revisados de Bethesda (24) donde se ampliaban los criterios anteriores.

La Sensibilidad de los criterios de Amsterdam II es de 87%, 62%, 38% y 48% para identificar pacientes con variantes patogénicas germinales en los genes MLH1, MSH2, PMS2 y MSH6 respectivamente; mientras que la Sensibilidad para los criterios Revisados de Bethesda para identificar pacientes con SL es >94%, pero con una especificidad del 25% (3).
El valor predictivo de los criterios de Amsterdam y Bethesda alcanza el 50% y 20%, respectivamente, lo que hace necesario el uso de herramientas complementarias como la IMS e inmunohistoquímica IHQ para el diagnóstico y manejo adecuado de estos pacientes (5). Lindor y colaboradores describieron algunos pacientes, que a pesar de cumplir con los criterios de Amsterdam, no presentaban alteraciones en los genes del sistema MMR, incluso sus familiares presentaban una menor incidencia de CCR respecto a las familias con SL, y no existía un alto riesgo a desarrollar cáncer extracolónico, por lo que sugirió la denominación de "CCR familiar tipo X" (25). Actualmente se ha determinado que el 40% de los pacientes que cumplen los criterios de Amsterdam no presentan mutaciones germinales en estos genes (5).
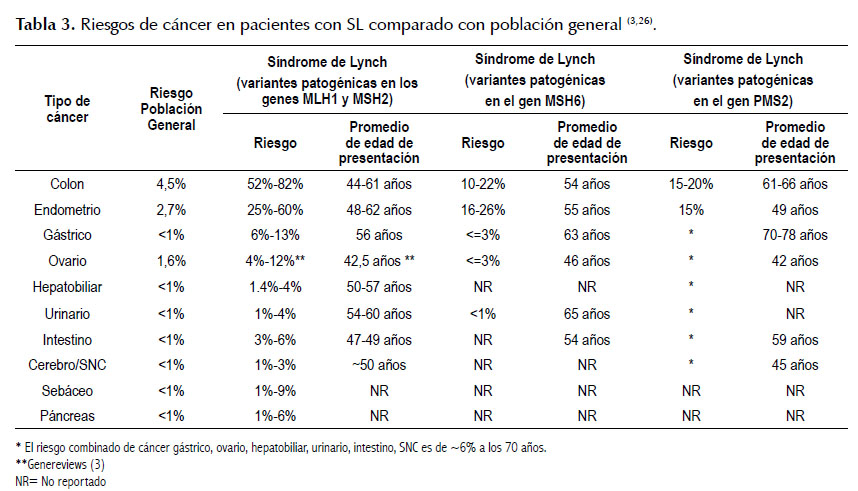
Riesgos de cáncer
El SL se caracteriza por conferir un alto riesgo a desarrollar CCR y otros tipos de cáncer como el cáncer de endometrio, ovario, estómago, intestino, hepatobiliar, tracto urinario, cerebro y piel comparado con la población general (26). Ver Tabla 3. También se ha descrito un alto riesgo a desarrollar cáncer de páncreas (8,6 veces mayor riesgo) y cáncer de próstata (2-5 veces mayor riesgo) (3). Existen estudios sobre otros tipos de cáncer como mama, sarcomas, carcinoma adrenocortical, sin embargo no se ha podido determinar la magnitud del riesgo asociado al SL.
Variantes del SL
1) Síndrome de Muir–Torre
Descrito inicialmente por Muir en el año 1967 (27) y por Torre en el año 1968 (28), este síndrome se caracteriza por la presencia de adenomas o carcinomas sebáceos, múltiples queratoacantomas y de neoplasias extracolónicas (3). Posteriormente en otros estudios (29–31), el Dr. Lynch y colaboradores definieron al Síndrome de Muir-Torre como una variante del SL.
2) Síndrome de Turcot
Se define por la presencia de CCR o adenomas colorrectales además de la presencia de tumores del Sistema Nervioso Central (SNC) y se clasifica en dos tipos según el gen alterado (3). Los pacientes con variantes patogénicas en el gen APC presentan pólipos y principalmente meduloblastomas. Mientras que en los pacientes con variantes patogénicas en los genes MMR, los tumores del SNC que se asocian frecuentemente son los glioblastomas los cuales presentan IMS (3).
3) Síndrome de deficiencia constitucional de MMR
Se han descrito casos de pacientes con variantes patogénicas en estado homocigoto en los genes MLH1, MSH2, MSH6 y PMS2, los cuales pueden presentar CCR o de intestino delgado en la segunda década de vida (3). Se presenta principalmente en pacientes pediátricos y se caracteriza además por el desarrollo de neoplasias hematológicas y del SNC, así como manchas café con leche, entre otros (32). Estos pacientes suelen tener historia familiar de SL, antecedente de consanguinidad entre progenitores y al menos un padre con hallazgos clínicos de SL (3).
Correlación genotipo-fenotipo
En el año 2001, fueron descritas algunas características anatomopatologícas como el CCR poco diferenciado, mucinoso, células anillo de sello, infiltración linfocítica peritumoral, reacción Crohn-like e infiltración linfocitaria incrementada en pacientes cuyos tumores presentaban IMS (33), siendo estos hallazgos utilizados hasta la actualidad como marcadores de probables casos.
De acuerdo al tipo de variante patogénica identificada en los genes MMR, se han identificado distintos fenotipos del SL (5). Las variantes patogénicas en el gen MSH2 son más frecuentes en pacientes con neoplasias extracolónicas y en el síndrome de Muir-Torre, mientras que las variantes patogénicas en el gen MLH1 son más frecuentes en pacientes con CCR (3). Las variantes patogénicas en el gen MSH6 se relacionan principalmente con cáncer de endometrio y de presentación más tardía comparado con pacientes con variantes en los otros genes MMR (3). Se ha sugerido que las variantes patogénicas en el gen PMS2 conllevan a presentar un fenotipo intermedio, de menor riesgo a desarrollar cáncer, así como ser de presentación tardía (5).
Las deleciones germinales en el gen EPCAM resultan en el silenciamiento del gen MSH2, asociándose a un riesgo incrementado a desarrollar CCR e incluso se ha propuesto que el riesgo a desarrollar neoplasias extracolónicas depende de la extensión de la deleción (3).
Penetrancia
La penetrancia en SL es incompleta (3), es decir que el paciente que posea una variante patogénica en alguno de los genes MMR, tiene una probabilidad menor al 100% de desarrollar algún cáncer relacionado al SL, por lo tanto, algunos individuos podrían nunca desarrollar cáncer.
Anticipación
Se ha descrito casos de hijos con cáncer desarrollados a una edad menor comparado con el cáncer desarrollado en los padres, sin embargo estos estudios no han podido ser reproducidos para corroborar la existencia de anticipación.
Diagnóstico diferencial
1) Síndrome de Lynch-like Pacientes con CCR cuyo tumor exhibe IMS, pero no poseen una variante patogénica germinal en los genes MMR (34).
2) Cáncer colorrectal familiar Corresponden a familias con historia de CCR en los cuales el análisis de IMS resulta estable, además de no existir un riesgo incrementado para neoplasias extracolónicas como las asociadas al SL (3). Estos casos se han asociado múltiples genes, alelos de baja penetrancia y factores ambientales que podrían contribuir con su desarrollo.
3) Poliposis adenomatosa familiar atenuada Es de una presentación intermedia y más tardía que la poliposis adenomatosa familiar clásica (gen APC), el cual se asocia a la presencia de pólipos colónicos (menor de 100). Usualmente los pólipos y CCR asociados no presentan IMS (3).
4) Poliposis asociada al gen MUTYH Esta condición genética se debe a variantes patogénicas bialélicas en el gen MUTYH, donde aproximadamente el 30% de casos corresponden a individuos con 15-100 pólipos o que poseen historia familiar de CCR en ausencia de múltiples pólipos (3).
5) Síndromes de poliposis hamartomatosa Se incluyen a diversas condiciones asociadas a un riesgo a desarrollar pólipos hamartomatosos y CCR, las cuales se diferencian por las manifestaciones extracolónicas principalmente (3).
- Síndrome de poliposis juvenil (genes SMAD4 y BMPR1A)
- Síndrome de Peutz-Jeghers (gen STK11)
- Síndromes hamartomatosos asociados al gen PTEN (Síndrome de Cowden y Síndrome de BannayanRiley-Ruvalcaba)
5) Cáncer gástrico difuso hereditario Caracterizado por la presencia de cáncer gástrico difuso (gen CDH1), anillo de sello, en pacientes jóvenes, donde las mujeres presentan además un alto riesgo a desarrollar cáncer de mama lobulillar (3).
6) Síndrome de cáncer de mama/ovario hereditario Debido a variantes patogénicas en los genes BRCA1 y BRCA2, este síndrome debe considerarse como diagnóstico diferencial si existe historia familiar de cáncer de ovario (3).
MÉTODOS DE DIAGNÓSTICO DEL SÍNDROME DE LYNCH
Inestabilidad de Microsatélites e Inmunohistoquímica
En el año 1993, se demostró que los pacientes con SL poseían una alteración molecular denominada Inestabilidad de Microsatélites (IMS) (35), la cual era consecuencia de un defecto en la reparación de los errores de la replicación del ADN.
Los microsatélites son repeticiones de uno a seis nucleótidos que se localizan en secuencias no codificantes a lo largo del genoma (10). Durante el proceso de replicación del ADN, suelen ocurrir errores en estas regiones del genoma, los cuales son identificados y corregidos por mecanismos de reparación postreplicativos del ADN (5). Cuando uno de los genes que codifican para las proteínas del sistema de reparación tiene inactivados sus dos alelos, los errores de la replicación del ADN se acumulan en estos segmentos de repeticiones, debido a que existe una ineficiencia en la actividad reparadora, a lo que se le denomina IMS (36). El estudio de la IMS nos permite conocer, indirectamente, que el sistema de reparación de los errores de la replicación del ADN está fallando tras evidenciarse longitudes alteradas de las repeticiones en tándem ubicadas en las secuencias microsatélites en el ADN tumoral (3).
Aproximadamente el 3% de todos los CCR corresponden al SL y de ellos el 90% presentan IMS (37). Sin embargo la IMS no es exclusiva del SL, ya que existen casos de CCR esporádicos con IMS debidos a la inactivación no hereditaria del sistema MMR, causado por la metilación del promotor del gen MLH1, lo que causa el silenciamiento de la expresión del gen, presentándose principalmente en personas mayores de 50 años (38). En estos casos, se requieren de estudios de metilación para definir la etiología esporádica y diferenciarlos de los casos hereditarios debidos a mutaciones germinales en el gen MLH1. Además existen casos de epimutación hereditaria o constitucional, que corresponden a pacientes con hipermetilación del gen MLH1 en tejido no tumoral (39).
El "panel Bethesda" de cinco marcadores para el estudio de la IMS compara el tejido tumoral con el tejido no tumoral (usualmente sangre) del paciente con sospecha de SL (5). Los marcadores que se utilizan son dos repeticiones de mononucleótidos (BAT25 y BAT26) y tres repeticiones de dinucleótidos (D2S123, D5S346 y D17S250), que permiten clasificar a los casos en IMS-alta cuando dos o más marcadores muestran diferencias en sus patrones en el tejido tumoral comparado con el no tumoral, en IMS-baja cuando uno de los marcadores muestra diferencias, y en Estable (EMS) cuando ninguno de los marcadores estudiados presenta alteraciones (5).
La IHQ evalúa la expresión de las proteínas MLH1, MSH2, MSH6 y PMS2 en el tejido tumoral. Dependiendo de la proteína que no se exprese en el tumor se podrá inferir el gen que podría poseer una alteración y de esta forma dirigir el secuenciamiento génico para la búsqueda de variantes patogénicas a nivel germinal (5). Las proteínas involucradas en la reparación de los errores en la replicación del ADN funcionan en colaboración unas con las otras, siendo así que las proteínas MLH1 y MSH2 interactúan con un segundo grupo de proteínas que incluye a PMS2 y MSH6 respectivamente (40). Este segundo grupo depende del primero, por tanto, cuando exista una variante patogénica germinal en el gen MLH1, se perderá la expresión de la proteína MLH1 y también de PMS2; del mismo modo, si existe una variante patogénica en el gen MSH2 habrá una pérdida de la expresión de la proteína MSH2 y de MSH6 (36). Sin embargo, cuando existan variantes patogénicas germinales en los genes PMS2 o MSH6, habrá una pérdida de expresión sólo de las proteínas PMS2 o MSH6 respectivamente (36,41).
La principal ventaja de la IHQ es su efectividad para detectar tumores resultantes de la deficiencia MMR con una sensibilidad del 92% (3) y además porque esta metodología se encuentra disponible en muchos centros hospitalarios. Sin embargo, posee limitaciones, como la variabilidad en la fijación del tejido que puede resultar en patrones de tinción débiles o equívocos, así como la necesidad de tener un adecuado tamaño tumoral para el análisis.
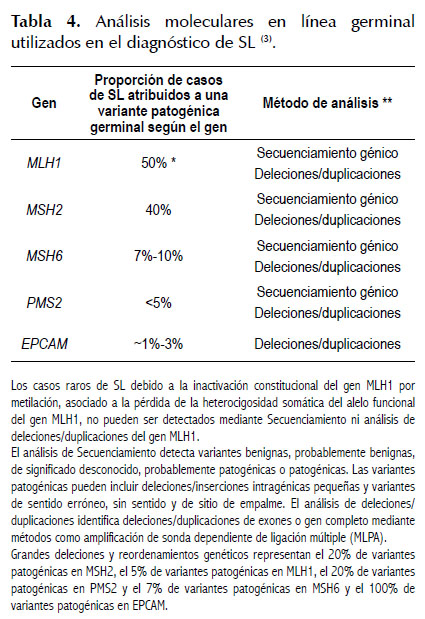
Análisis molecular de los genes MMR a nivel germinal
El SL se debe a variantes patogénicas germinales en los genes del sistema MMR, llamados MLH1 (mutL homolog 1, colon cancer, nonpolyposis type 2) (42,43), MSH2 (mutS homolog 2, colon cancer, nonpolyposis 1) (40,44) y MSH6 (mutS homolog 6) (45,46) y el menos frecuente PMS2 (postmeiotic segregation increased 2) (47). También se debe a deleciones en el gen EPCAM en el 1% de los casos (3), el cual no es un gen del sistema MMR. Ver Tabla 4.
El desarrollo del cáncer en estos pacientes está basado en la hipótesis del segundo hit de Knudson (8), donde la variante patogénica germinal sería el primer hit, confiriendo una susceptibilidad hereditaria a desarrollar el cáncer y posteriormente se generaría un segundo hit, ya sea una deleción o una variante patogénica puntual adquirida, con pérdida de función del alelo normal a nivel somático en los tejidos vulnerables.
El diagnóstico definitivo del SL se establece mediante la identificación de alguna variante patogénica en los genes MMR o en el gen EPCAM, mediante el secuenciamiento o el análisis de deleciones/duplicaciones de estos genes.
Modelos matemáticos predictivos
Actualmente existen modelos matemáticos predictivos que tras ingresar los datos de la historia personal y familiar de los pacientes con sospecha de SL, principalmente aquellos que hayan desarrollado CCR, permiten calcular la probabilidad de que este sea portador de una variante patogénica en los genes MMR (5). Sin embargo, estos modelos tienen sus limitaciones para determinar la probabilidad de portar mutaciones germinales en aquellos pacientes que hayan desarrollado exclusivamente neoplasias extracolónicas. Presentamos la información más relevante sobre los modelos matemáticos predictivos aplicados en SL. Ver Tabla 5.

ALGORITMO DIAGNÓSTICO
El diagnóstico del SL se realiza a partir de los datos clínicos del paciente, en base a sus antecedentes personales y familiares, criterios clínicos, análisis por IMS y/o IHQ de los tumores y estudios moleculares en búsqueda de variantes patogénicas germinales en los genes MMR. Ver Tabla 6, Figura 2 y Tabla 7.

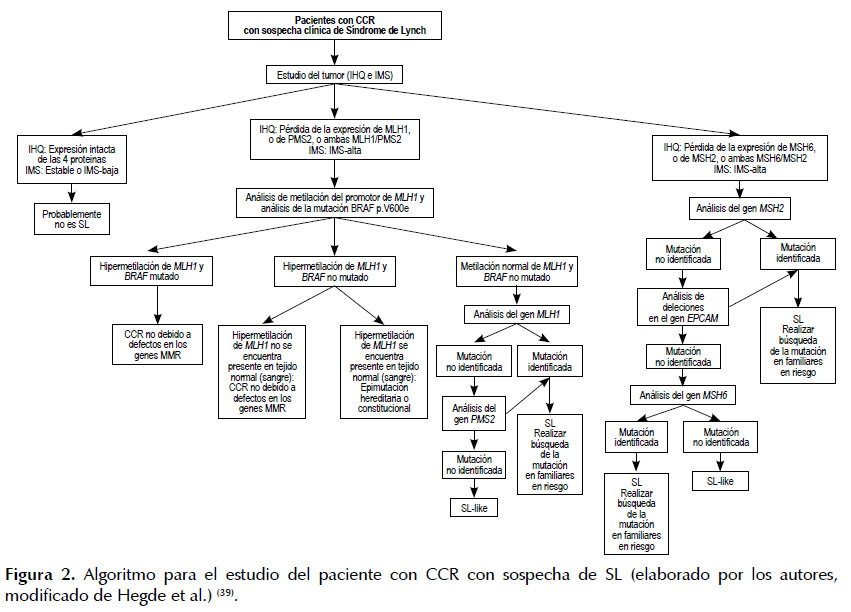
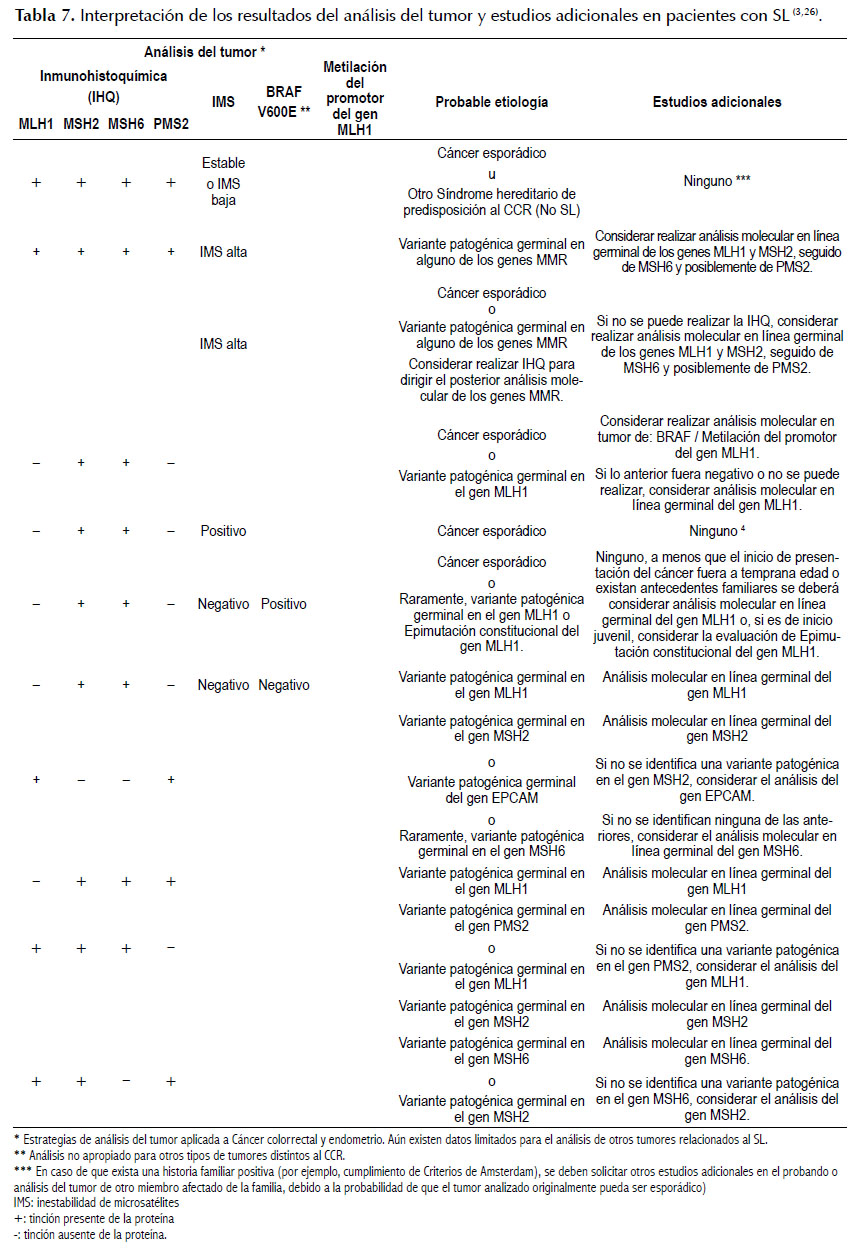
1. Estudio del tumor: El análisis de la pieza tumoral mediante estudios de biología molecular, permite identificar si el paciente posee un tumor de tipo esporádico o hereditario, además de poder influir en el manejo médico a través de las terapias dirigidas.
a. Análisis molecular de IMS (marcadores del panel Bethesda) y/o IHQ (evaluación de la expresión de las 4 proteínas). Estos estudios se realizan en pacientes que posean CCR y/u otros tipos de tumores relacionados al SL, así como edad temprana de presentación o historia familiar positiva. La presencia de IMS en el tumor, sin otro hallazgo clínico en el paciente, no es suficiente para diagnóstico de SL, debido a que el 1015% de casos de CCR esporádico pueden presentar IMS. El estudio de IHQ ayuda a identificar la proteína defectuosa infiriendo que gen podría poseer una variante patogénica germinal.
b. Análisis molecular de metilación del gen MLH1 y variantes patogénicas somáticas en el gen BRAF (p.V600E). Ambos en el tumor, para ayudar a identificar aquellos tumores que probablemente sean esporádicos más que hereditarios.
Continuando con el algoritmo diagnóstico, si un paciente posee un resultado de IMS-alta se establecerá la sospecha de ser un caso hereditario y el análisis de IHQ permitirá orientar cuál de los genes MMR podría presentar una variante patogénica germinal. El siguiente incluye el análisis del ADN del paciente, en sangre periférica, por secuenciamiento génico dirigido a los genes MMR.
2. Estudio en sangre periférica:
a. Estudio genético molecular de los genes MMR. Permite identificar una variante patogénica cuando los hallazgos clínicos y del tumor son consistentes con SL. Este estudio incluye el secuenciamiento génico y análisis de deleciones/duplicaciones de los genes MMR. El secuenciamiento génico se realiza principalmente a través del análisis de un panel multigenes (MLH1, MSH2, MSH6, PMS2 y EPCAM) el cuál se realiza mediante la metodología de secuenciamiento de próxima generación (NGS, next generation sequencing). La confirmación de las variantes patogénicas identificadas mediante este panel, se realiza a través del secuenciamiento tipo Sanger.
b. En los casos en que el tejido tumoral no se encuentre disponible, se podrá realizar el análisis en sangre periférica como primer paso. Si el miembro de la familia afectado por cáncer hubiese fallecido y no se cuente con el tejido tumoral, se podrá realizar el análisis en un familiar sano teniendo en cuenta que la probabilidad de que se identifique una alteración genética dependerá del grado de parentesco que posea con el miembro afectado fallecido.
ASESORÍA GENÉTICA Y CONTROL DE RIESGOS
Los genes MMR se heredan bajo un patrón de herencia autosómico dominante, por lo que la mayoría de pacientes con SL poseen un progenitor afectado. Sin embargo, debido a la penetrancia incompleta y la edad variable de presentación del cáncer, así como a las medidas de reducción de riesgo mediante cirugías profilácticas, o muerte a temprana edad, no todos los pacientes con variantes patogénicas en los genes MMR tienen un progenitor con cáncer. Cada hijo de un paciente con SL tiene el 50% de probabilidad de heredar la variante patogénica.
Sabemos que la evaluación del riesgo del cáncer basado en la historia personal y familiar del paciente puede cambiar significativamente una vez detectada la variante patogénica en la familia y esta es rastreada en el resto de sus familiares. Conocer si una persona es portador o no de una variante que predispone el desarrollo del cáncer es un hecho fundamental en la asesoría genética de los pacientes en riesgo.
Todo paciente con cáncer colorrectal o del especto del SL que cumpla con los criterios clínicos de sospecha, debe ser referido a una evaluación por un médico genetista para recibir la asesoría genética correspondiente y determinar si el caso es hereditario o esporádico. Incluso, estableciéndose que el origen es hereditario, es necesario tener en cuenta que existen otros síndromes de predisposición genética al cáncer colorrectal que no corresponden a SL, tales como Síndrome de Cowden, poliposis colónicas familiares, etc, por lo que la evaluación y asesoría genética pretest será fundamental para continuar con el estudio del caso.
La asesoría genética en los pacientes con SL incluye una evaluación previa y posterior a los estudios genéticos y permite: 1) conocer los antecedentes personales y familiares oncológicos del individuo y representarlo mediante un árbol genealógico; 2) establecer el diagnóstico de SL según criterios clínicos-genéticos; 3) solicitar estudios genéticos confirmatorios e informar al paciente los posibles resultados; y 4) explicar de forma objetiva los resultados, así como las causas genéticas del SL y las implicancias tanto en el paciente como en sus familiares.
Existen diversos reportes en familias con SL (48-51) que mencionan los beneficios en el manejo y seguimiento con colonoscopía en los pacientes portadores de mutaciones germinales, como una disminución del 62% en la incidencia de CCR y disminución del 65% en la mortalidad global. Las recomendaciones actuales son las de realizar colonoscopias anuales desde los 25 años en aquellos portadores de mutaciones germinales (5) y en los familiares en riesgo, debido a que la presentación del CCR en los casos de SL se desarrolla a edades más tempranas y posee una carcinogénesis más acelerada comparada con los casos esporádicos.
Además del control de riesgos de CCR, se debe tener en cuenta el control ginecológico por la alta incidencia de cáncer de endometrio, principalmente, y cáncer de ovario en el SL. El manejo se basa en el examen clínico, ecografía transvaginal, aspirado endometrial y CA125 de forma anual con una edad de inicio de 30 años de edad en las mujeres portadoras de una variante patogénica germinal (5), sin embargo la Guía Clínica de la Sociedad Americana de Gastroenterología (SAG) en el año 2015 presentó esta recomendación con un nivel de tipo condicional y con muy baja calidad de la evidencia (52). Ver Tabla 8.
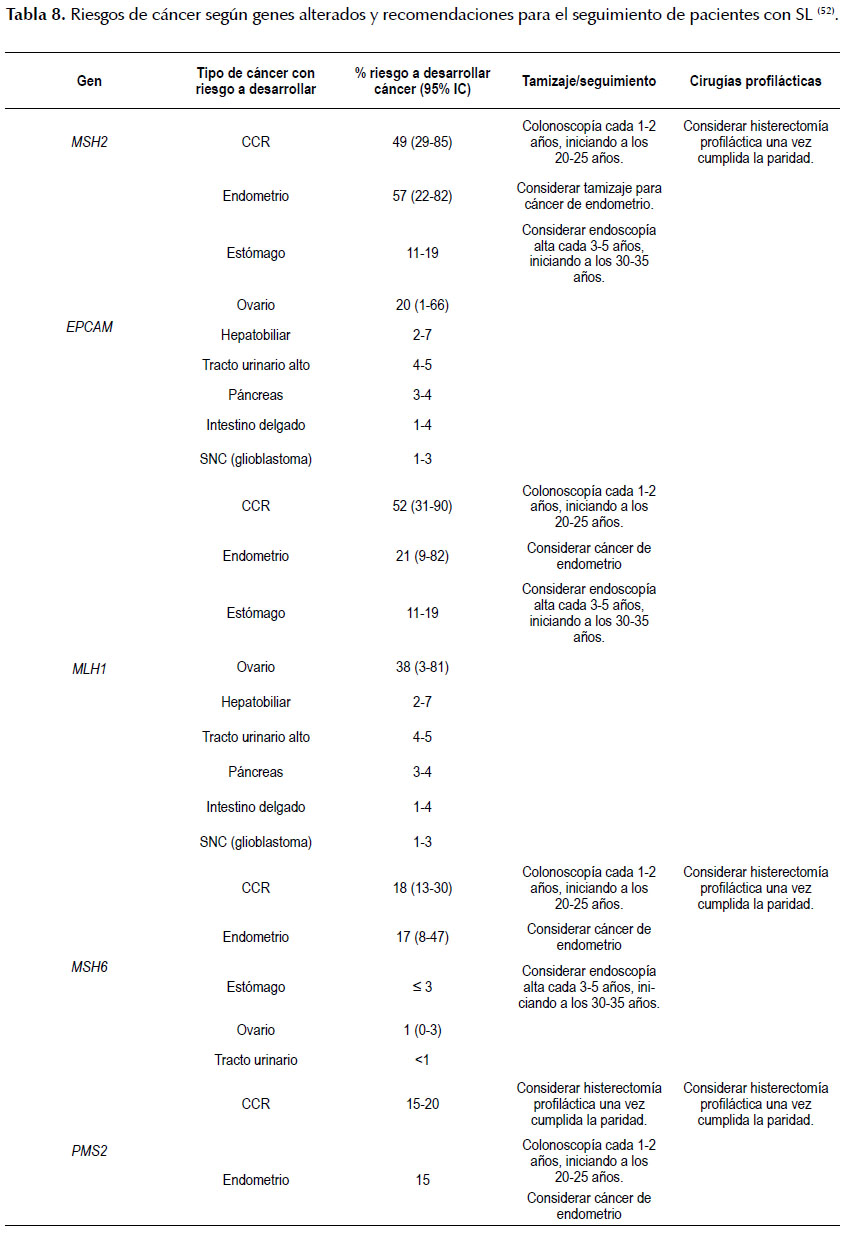
Así mismo, ante la recomendación de realizar histerectomía y salpingooforectomía bilateral profiláctica en mujeres que hayan cumplido deseo de paridad, algunas publicaciones científicas se pronuncian a favor (53,54), mientras que la Guía Clínica de la SAG (52) indica que estas recomendaciones tendrían un nivel de tipo condicional ya que poseen una baja calidad de la evidencia. Además, se ha recomendado realizar el control anual con ecografía pélvica y análisis de orina en los pacientes portadores de variantes patogénicas germinales que tengan historia familiar de cáncer de uréter o pelvis renal, así mismo la endoscopía alta se recomienda en los portadores de variantes patogénicas con antecedentes familiares de cáncer gástrico o duodeno (36), pero también es discutido en la Guía Clínica de la SAG (52) bajo un nivel de recomendación condicional.
Parte del manejo de control de riesgos en los portadores de variantes patogénicas incluyen las cirugías profilácticas, siendo en el caso de CCR una colectomía subtotal que aumenta la expectativa de vida hasta en 2 años en comparación a la resección segmentaria del tumor (36). Esto se sustenta a que en 1996 (55), el Dr. Lynch demostró que la colectomía profiláctica disminuía los riesgos en los portadores de variantes patogénicas germinales o en aquellos que no deseaban realizar un seguimiento con colonoscopías anuales. La Guía Clínica de la SAG (52) presenta a esta recomendación como condicional y con moderada calidad de la evidencia. Ver Tabla 9.

Hasta la actualidad existen dificultades en la identificación de variantes patogénicas relacionadas al SL, debido a que probablemente existan otros genes causantes aun no estudiados, además de los diversos fenotipos que pueden presentarse. Por tal motivo, el diagnóstico de los pacientes con SL requiere de un equipo multidisciplinario que conozca las bases genéticas y moleculares de este síndrome y maneje de forma integral al paciente y familiares en riesgo, haciendo énfasis en la historia personal y familiar del paciente con CCR y/u otra neoplasia extracolónica asociada a este síndrome. La identificación oportuna de estas familias determinará que se realice la asesoría genética al paciente y familiares en riesgo, estableciendo medidas de seguimiento y de prevención de forma personalizada, basada en las guías clínicas e información científica relevante, a fin de evitar la morbimortalidad por cáncer.
Conflicto de intereses: Los autores declaran no tener conflicto de intereses.
REFERENCIAS BIBLIOGRÁFICAS
1. McKusick VA. Lynch Syndrome I [Internet]. Baltimore, MD: OMIM; c 1966-2018 [citado 4 de agosto de 2018]. Disponible en: http://omim.org/entry/120435 [ Links ]
2. Lynch HT, Lynch JF, Shaw TG. Hereditary gastrointestinal cancer syndromes. Gastrointest Cancer Res GCR. 2011;4(4 Suppl 1):S9-17. [ Links ]
3. Kohlmann W, Gruber SB. Lynch Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. SourceGeneReviews® [Internet]. Seattle (WA): University of Washington; c1993-2018. 2004 Feb 5 [updated 2018 Apr 12].
4. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919-32. [ Links ]
5. Bandres F, Urioste M. Planteamientos básicos del cáncer hereditario: principales síndromes. Madrid: Fundación Tejerina; 2011. [ Links ]
6. Thorson AG, Knezetic JA, Lynch HT. A century of progress in hereditary nonpolyposis colorectal cancer (Lynch syndrome). Dis Colon Rectum. 1999;42(1):1-9. [ Links ]
7. Classics in oncology. Heredity with reference to carcinoma as shown by the study of the cases examined in the pathological laboratory of the University of Michigan, 1895- 1913. By Aldred Scott Warthin. 1913. CA Cancer J Clin. 1985;35(6):348-59. [ Links ]
8. Lynch HT, Hitchins MP, Shaw TG, Lynch JF, Roy H. Historical Aspects of Lynch Syndrome. In: Rodriguez-Bigas MA, Cutait R, Lynch PM, Tomlinson I, Vasen HFA. Hereditary Colorectal Cancer. New York: Springer US; 2010. p. 15-42. [ Links ]
9. Lynch HT, Shaw MW, Magnuson CW, Larsen AL, Krush AJ. Hereditary factors in cancer. Study of two large midwestern kindreds. Arch Intern Med. 1966;117(2):206-12. [ Links ]
10. Sánchez, A, Benavides, M, Blanco, I, Urioste, M. Cáncer Hereditario. Madrid: Sociedad Española de Oncología Médica; 2006. [ Links ]
11. Lynch HT, Krush AJ. Cancer family «G» revisited: 1895-1970. Cancer. 1971;27(6):1505-11. [ Links ]
12. Lynch HT. Family information service and hereditary cancer. Cancer. 2001;91(4):625-8. [ Links ]
13. Lynch PM, Lynch HT, Harris RE. Hereditary proximal colonic cancer. Dis Colon Rectum. 1977;20(8):661-8. [ Links ]
14. Watson P, Lynch HT. Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer. 1993;71(3):677-85. [ Links ]
15. Boland CR, Troncale FJ. Familial colonic cancer without antecedent polyposis. Ann Intern Med. 1984;100(5):700-1. [ Links ]
16. Boland CR. Evolution of the nomenclature for the hereditary colorectal cancer syndromes. Fam Cancer. 2005;4(3):211-8. [ Links ]
17. Bansidhar BJ, Silinsky J. History and pathogenesis of lynch syndrome. Clin Colon Rectal Surg. 2012;25(2):63-6. [ Links ]
18. Sameer AS, Nissar S, Fatima K. Mismatch repair pathway: molecules, functions, and role in colorectal carcinogenesis. Eur J Cancer Prev. 2014;23(4):246-57. [ Links ]
19. Martín-López JV, Fishel R. The mechanism of mismatch repair and the functional analysis of mismatch repair defects in Lynch syndrome. Fam Cancer. 2013;12(2):159-68. [ Links ]
20. Martin SA, Lord CJ, Ashworth A. Therapeutic targeting of the DNA mismatch repair pathway. Clin Cancer Res. 2010;16(21):5107-13. [ Links ]
21. Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum. 1991;34(5):424-5. [ Links ]
22. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248-57. [ Links ]
23. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116(6):1453-6. [ Links ]
24. Laghi L, Bianchi P, Roncalli M, Malesci A. Re: Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(18):1402-3; author reply 1403-4. [ Links ]
25. Lindor NM, Rabe K, Petersen GM, Haile R, Casey G, Baron J, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA. 2005;293(16):1979-85. [ Links ]
26. National Comprehensive Cancer Network [Internet]. Plymouth Meeting, PA: NCCN; c2018 [citado 4 de agosto de 2018]. Disponible en: https://www.nccn.org/store/login/ login.aspx?ReturnURL=https://www.nccn.org/professionals/ physician_gls/pdf/genetics_colon.pdf [ Links ]
27. Muir EG, Bell AJ, Barlow KA. Multiple primary carcinomata of the colon, duodenum, and larynx associated with keratoacanthomata of the face. Br J Surg. 1967;54(3):191-5. [ Links ]
28. Torre D. Multiple sebaceous tumors. Arch Dermatol. 1968;98(5):549-51. [ Links ]
29. Lynch HT, Lynch PM, Pester J, Fusaro RM. The cancer family syndrome. Rare cutaneous phenotypic linkage of Torre’s syndrome. Arch Intern Med. 1981;141(5):607-11.
30. Lynch HT, Fusaro RM, Roberts L, Voorhees GJ, Lynch JF. Muir-Torre syndrome in several members of a family with a variant of the Cancer Family Syndrome. Br J Dermatol. 1985;113(3):295-301. [ Links ]
31. Fusaro RM, Lemon SJ, Lynch HT. Muir-Torre syndrome and defective DNA mismatch repair genes. J Am Acad Dermatol. 1996;35(3 Pt 1):493-4. [ Links ]
32. Bandipalliam P. Syndrome of early onset colon cancers, hematologic malignancies & features of neurofibromatosis in HNPCC families with homozygous mismatch repair gene mutations. Fam Cancer. 2005;4(4):323-33. [ Links ]
33. Smyrk TC, Watson P, Kaul K, Lynch HT. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer. 2001;91(12):2417-22. [ Links ]
34. Kang SY, Park CK, Chang DK, Kim JW, Son HJ, Cho YB, et al. Lynch-like syndrome: characterization and comparison with EPCAM deletion carriers. Int J Cancer. 2015;136(7):1568-78. [ Links ]
35. Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363(6429):558-61. [ Links ]
36. Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76(1):1-18. [ Links ]
37. Boland CR. Clinical uses of microsatellite instability testing in colorectal cancer: an ongoing challenge. J Clin Oncol Off J Am Soc Clin Oncol. 2007;25(7):754-6. [ Links ]
38. Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A. 1998;95(15):8698-702. [ Links ]
39. Hegde M, Ferber M, Mao R, Samowitz W, Ganguly A, Working Group of the American College of Medical Genetics and Genomics (ACMG) Laboratory Quality Assurance Committee. ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYHassociated polyposis). Genet Med Off J Am Coll Med Genet. 2014;16(1):101-16. [ Links ]
40. Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75(5):1027-38. [ Links ]
41. Boland CR, Koi M, Chang DK, Carethers JM. The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch syndrome: from bench to bedside. Fam Cancer. 2008;7(1):41-52. [ Links ]
42. Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary nonpolyposis colon cancer. Nature. 1994;368(6468):258-61. [ Links ]
43. Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, et al. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263(5153):1625-9. [ Links ]
44. Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75(6):1215-25. [ Links ]
45. Akiyama Y, Sato H, Yamada T, Nagasaki H, Tsuchiya A, Abe R, et al. Germ-line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res. 1997;57(18):3920-3. [ Links ]
46. Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Yasuno M, et al. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet. 1997;17(3):271-2. [ Links ]
47. Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, Ruben SM, et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature. 1994;371(6492):75-80. [ Links ]
48. Douglas JA, Gruber SB, Meister KA, Bonner J, Watson P, Krush AJ, et al. History and molecular genetics of Lynch syndrome in family G: a century later. JAMA. 2005;294(17):2195-202. [ Links ]
49. Järvinen HJ, Mecklin JP, Sistonen P. Screening reduces colorectal cancer rate in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 1995;108(5):1405-11. [ Links ]
50. Vasen HF, van Ballegooijen M, Buskens E, Kleibeuker JK, Taal BG, Griffioen G, et al. A cost-effectiveness analysis of colorectal screening of hereditary nonpolyposis colorectal carcinoma gene carriers. Cancer. 1998;82(9):1632-7. [ Links ]
51. Järvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomäki P, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118(5):829-34. [ Links ]
52. Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW, et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110(2):223-262; quiz 263. [ Links ]
53. Schmeler KM, Lynch HT, Chen L, Munsell MF, Soliman PT, Clark MB, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med. 2006;354(3):261-9. [ Links ]
54. Lindor NM, Petersen GM, Hadley DW, Kinney AY, Miesfeldt S, Lu KH, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA. 2006;296(12):1507-17. [ Links ]
55. Lynch HT. Is there a role for prophylactic subtotal colectomy among hereditary nonpolyposis colorectal cancer germline mutation carriers? Dis Colon Rectum. 1996;39(1):109-10. [ Links ]
Correspondencia:
María del Carmen Castro-Mujica
E-mail: mc.castro.mujica@gmail.com
Recibido: 04/08/2018
Aprobado: 22/10/2018

