










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Gastroenterología del Perú
versión impresa ISSN 1022-5129
Rev. gastroenterol. Perú vol.39 no.3 Lima jul./set. 2019
REPORTE DE CASO
Meduloblastoma y meningioma recidivante asociados a poliposis colónica: una presentación inusual del síndrome de Turcot
Meduloblastoma and recurrent meningioma in association with colonic polyposis: an unusual presentation of Turcot syndrome
Carlos Barreda Costa1a, Lang Chu Revollar1a, Angella Herrera Alzamora1b
1 Servicio de Gastroenterología, Clínica Ricardo Palma. Lima, Peru.
a Médico gastroenterólogo, b Médico cirujano
RESUMEN
El síndrome de Turcot es un desorden genético caracterizado por la asociación de tumores primarios neuroepiteliales del sistema nervioso central y poliposis adenomatosa del colon. Se describen dos variedades. En el tipo I los tumores suelen ser glioblastomas y se asocian a un síndrome de Lynch o cáncer colorectal hereditario no polipósico. En el tipo II predominan los meduloblastomas y se asocian a poliposis múltiple familiar, ya sea la forma clásica o atenuada. El presente caso debutó a los 7 años de edad con un meduloblastoma que logró ser curado, pero 20 años después desarrolla un meningioma cerebral recidivante. A los 36 años presenta anemia por sangrado digestivo y se descubre a la colonoscopía una poliposis adenomatosa del colon, con displasia de alto grado. Hasta donde conocemos es el primer caso de síndrome de Turcot que se reporta en nuestro país.
Palabras clave: Síndrome de Turcot; Meduloblastoma; Meningioma (fuente: DeCS BIREME).
ABSTRACT
Turcot syndrome is an association of primary neuroepithelial tumors of the central nervous system with adenomatous polyposis coli. It is a genetic disorder, with two forms; In type I, glioblastomas are usually associated with hereditary nonpolyposis colorectal cancer (HNPC or Lynch). In Type II, medulloblastomas are often associated with familial adenomatous polyposis coli (classical or attenuated). This patient had a medulloblastoma at seven years of age, then 20 years later developed a meningioma which recurred several times. At 36 years old he presented with anemia after digestive bleeding, and an adenomatous polyposis coli with high grade dysplasia was found at colonoscopy. As far as we know, this is the first case of Turcot syndrome described in our country.
Keywords: Turcot síndrome; Medulloblastoma; Meningioma (source: MeSH NLM).
INTRODUCCIÓN
El síndrome de Turcot (ST) es un síndrome hereditario muy raro, del cual solo se han reportado unos 177 casos en la literatura mundial, pero varios de ellos no tienen confirmación histopatológica y otros son recopilaciones que incluyen casos ya publicados, por lo cual el número real parece ser menor. En el Perú, revisando la bibliografía a nuestro alcance, no hemos encontrado ningún caso publicado.
Se caracteriza por la asociación de tumores primarios neuroepiteliales del sistema nervioso central (SNC) y poliposis adenomatosa colorrectal (1). Los tumores del SNC suelen aparecer antes, en la primera o segunda década de la vida, e incluyen meduloblastomas, gliomas o glioblastomas, aunque se han descrito otros, como astrocitomas. Si el paciente sobrevive a estas neoplasias, comienza a desarrollar múltiples adenomas del colon, los cuales poseen una alta tendencia a la malignización, por lo cual muchos de estos pacientes desarrollan cáncer de colon antes de llegar a la tercera o cuarta década de la vida (1,2).
Los patrones de herencia del ST han sido difíciles de determinar; inicialmente se le consideraba como una variante de la poliposis múltiple familiar (PMF), pero luego se ha determinado que es una enfermedad diferente, con alteraciones genéticas propias y que existen al menos dos subtipos (3).
El denominado tipo I, que incluye a los dos casos descritos por Turcot en 1959 (4) se asocia a mutaciones en los genes MMR, reparadores de la cadena de DNA, como fue demostrado años después por Hamilton en 1995 (5). Estos pacientes presentan un fenotipo semejante al Síndrome de Lynch, con un número limitado de adenomas colónicos, que tienen tendencia a ser planos y a localizarse en el colon derecho.
Por otro lado, el tipo II presenta mutación en los genes APC (Adenomatous poliposis coli) y las lesiones colónicas se asemejan más a las encontradas en la poliposis múltiple familiar (PMF) aunque el número de adenomas suele ser menor de 100, por lo cual se enmarcarían mejor en la denominada PMF atenuada (2,6).
El caso que presentamos es inusual, por haber desarrollado inicialmente un meduloblastoma y posteriormente un segundo tumor del SNC. Se han descrito casos de poliposis adenomatosa del colon asociada a tumores no neuroepiteliales del SNC, como menigiomas, tumores de hipófisis, craneofaringiomas, sarcomas y linfomas, pero el consenso general es que estos últimos tumores no formarían parte del ST, sino corresponderían a una patología coincidente (1,7).
CASO CLÍNICO
Paciente varón nacido de parto eutócico, que a los 7 años de edad desarrolló un cuadro de hipertensión endocraneana, con cefalea, vómitos y convulsiones. Se detectó por tomografías un tumor en la fosa posterior del cerebro, siendo sometido a cirugía en el Instituto Nacional de Enfermedades Neoplásicas (INEN). Se reseco la lesión, constatándose que se trataba de un meduloblastoma. Luego recibió quimioterapia y un ciclo de radioterapia cerebro espinal.
A los 27 años de edad vuelve a presentar cuadros convulsivos repetidos y debilidad muscular. Al realizar estudios de imágenes y se descubre un nuevo tumor cerebral (Figura 1). Es operado, esta vez en nuestra Institución, resecándose parcialmente un tumor que resultó ser un menigioma transicional y fibroso de grado histológico I, según el estudio anatomopatológico. El paciente experimentó sucesivas recurrencias de este tumor, siendo sometido a 6 intervenciones quirúrgicas a lo largo de los siguientes 9 años, a consecuencia de lo cual quedó con secuelas neurológicos que incluyen deterioro mental, hipoacusia, amaurosis y marcada inestabilidad para la marcha.
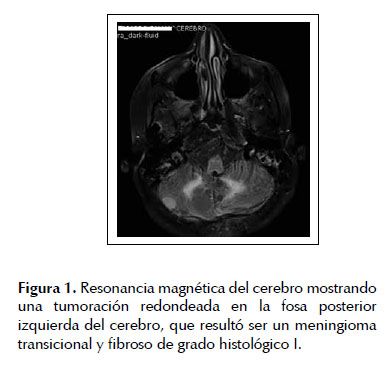
Unas semanas después de la última cirugía, a los 36 años, presenta leve sangrado rectal intermitente, seguido de palidez, evidenciándose una anemia con hemoglobina en 9,6 g/dl. Por este motivo es referido al servicio de gastroenterología.
No tenía antecedentes familiares de cáncer de colon, ni de tumores del SNC. Solo un abuelo paterno había presentado cáncer de próstata a edad avanzada. Sus dos padres y su único hermano estaban sanos, no habiendo sido sometidos nunca a exámenes endoscópicos del tracto digestivo.
Al examen clínico se encontró a un paciente despierto, pálido y algo adelgazado, con severa hipoacusia, que se expresaba con un lenguaje limitado a unos pocos monosílabos. Se notaba una evidente deformación de la bóveda craneana, como secuela de las múltiples cirugías a las que había sido sometido. No se palpaban tumoraciones en cabeza y cuello. Tórax y pulmones normales. El abdomen era blando, no doloroso, con ruidos intestinales normales. No había distensión, ni masas palpables. No signos de ascitis, ni edemas. Tenía dificultad para la marcha, con debilidad muscular generalizada y tono muscular disminuido.
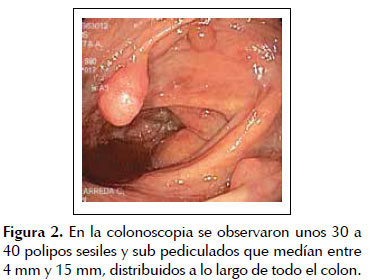
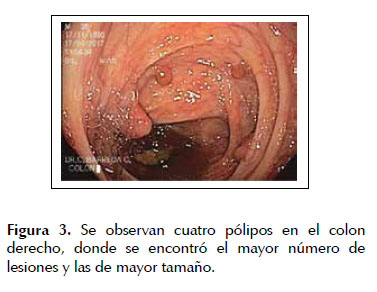
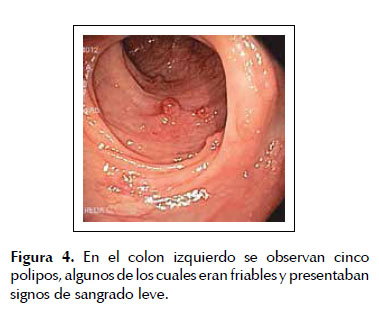
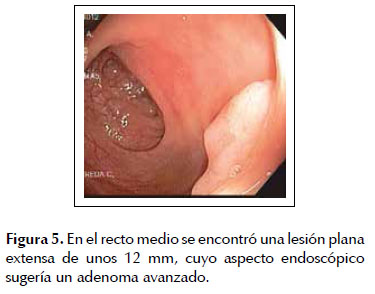
Se le realizó una colonoscopía evidenciándose un total de 30 a 40 pólipos, distribuidos a lo largo del colon y recto, con predominio del recto, colon transverso y ascendente. Las lesiones eran sésiles o sub pediculadas y de diversos tamaños, que iban entre unos 4 mm hasta unos 15 mm. Algunas eran friables y mostraban erosión en su superficie, con escasos restos hemáticos (Figuras 2, 3, 4 y 5). El paciente mostró poca tolerancia y falta de colaboración con el examen, a pesar de la sedación. Por este motivo se decidió tratar solo las lesiones de mayor tamaño, extirpándose con asa térmica un total de 5 pólipos, los cuales fueron rotulados y enviados a patología en frascos separados.
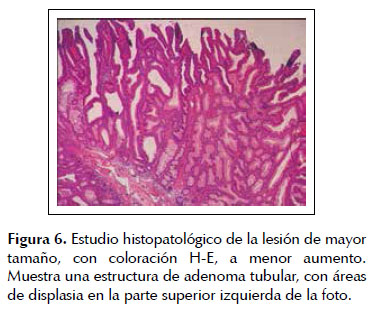
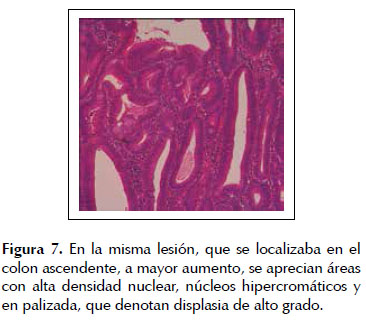
El examen histopatológico de los pólipos resecados mostró que se trataba de adenomas tubulares con displasia, de grado leve a moderado en 4 de ellas y de alto grado en la de mayor tamaño, que se localizaba en el colon ascendente (Figuras 6 y 7).
El paciente ha mantenido niveles de hemoglobina por encima de 11 g% y no ha presentado nuevos episodios de sangrado rectal visible después de la colonoscopía. Mantiene un seguimiento estricto por neurocirugía.
DISCUSIÓN
El diagnóstico clínico del síndrome de Turcot se sustenta en una combinación de poliposis adenomatosa múltiple del colon y tumores neuroepiteliales primarios del sistema nervioso central, del tipo meduloblastuma, gliomas, glioblastoma y otros menos comunes. Su sustrato genético es heterogéneo, y se han descrito dos subtipos, como se mencionó en la introducción; El tipo I exhibe herencia autosómica recesiva, con mutaciones del gen MMR y suele asociarse a glioblatomas, mientras que el Tipo II muestra herencia autosómica dominante con mutaciones del gen APC (Adenomatous polyposis coli) y se asocia más a meduloblastomas. Existen unos pocos casos en los que no se logra identificar el gen afectado, pero no se han reportado casos asociados a mutaciones del gen MUTYH (1,3).
El antecedente de cáncer o adenomas múltiples del colon, así como de tumores neuropepiteliales del SNC en familiares cercanos, sobre todo menores de 40 años, apoya fuertemente el diagnóstico de síndrome de Turcot, pero no suele estar presente en estos casos. La mayoría de pacientes afectados por este síndrome sufre severas secuelas neurológicas o muere por cáncer de colon a temprana edad y no llega a dejar descendencia. Se describe con mayor frecuencia afectación de los hermanos. En una revisión reciente publicada en esta revista, se encontró que el 46,5% de los casos catalogados como síndromes hereditarios con predisposición al cáncer colorrectal, eran casos aislados y no tenían otros familiares afectados. Como no se realizan en nuestro medio estudios de secuenciamiento genético y las muestras deben ser enviadas al extranjero, con un alto costo, el referido estudio recomienda usar criterios clínico-genéticos para identificar a estos pacientes (8). Otra revisión sobre el ST sostiene que un 25% de los casos observados constituyen mutaciones "de novo" y no se encuentran otros familiares afectados (9).
Por estas consideraciones, el caso que presentamos no puede ser excluido por la falta de antecedentes familiares de neoplasias, ya que cumple los criterios clínicos e histopatológicos para el diagnóstico de síndrome de Turcot. Presentó un meduloblastoma a temprana edad, seguido años después por un menigioma. No se ha descrito una asociación más allá de lo casual, entre estas dos neoplasias. Es posible que el tratamiento de radioterapia cerebro espinal que recibió nuestro paciente haya sido un factor condicionante del menigioma, lo cual ha sido descrito (2).
Su poliposis colónica recién se manifiesta a los 36 años de edad, con anemia secundaria a sangrado digestivo, y las lesiones ya tenían displasia de alto grado al momento del diagnóstico. Este tipo de presentación es muy común en estos casos (2,10,11). Por sus características nuestro caso puede enmarcarse en un síndrome de Turcot tipo II, asociado a una poliposis múltiple familiar de forma atenuada, por tener menos de 100 adenomas del colon.
Considerando su avanzado compromiso neurológico y el hecho de no haber presentado aún cáncer en los adenomas colónicos, se le ha recomendado un seguimiento con colonoscopías anuales, a fin de extirpar las lesiones de mayor riesgo, en lugar indicarle una colectomía total. Esto es lo que recomiendan algunos autores para el manejo de la PMF atenuada, ya que su riesgo de cáncer no es tan elevado como en la forma clásica (12). También se ha explicado a sus padres y su hermano la importancia de hacerse una colonoscopía, para prevenir el desarrollo de cáncer de colon, debido al alto riesgo genético que existe en estos casos. Por motivos de índole personal y cultural, hasta la fecha no han accedido.
En Latinoamérica han sido reportados cuatro casos del Síndrome de Turcot, el último de ellos en Colombia (13), pero no hemos encontrado ningún caso en el Perú, por lo que éste sería el primer caso publicado. En conclusión, creemos que es importante tener en consideración que esta rara enfermedad también puede presentarse en nuestro medio y debe ser reconocida. Los pacientes que han tenido en la infancia alguno de los tumores del SNC asociados a este síndrome, están en riesgo de presentarla y deben ser investigados en la segunda década de vida, o antes si presentan sangrado digestivo o anemia, a fin de detectar a tiempo las lesiones del colon e impedir su malignización.
Conflicto de intereses: ninguno.
Citar como: Barreda Costa C, Chu Revollar L, Herrera Alzamora A. Meduloblastoma y meningioma recidivante asociados a poliposis colónica: una presentación inusual del Síndrome de Turcot. Rev Gastroenterol Peru. 2019;39(3):280-3
REFERENCIAS BIBLIOGRÁFICAS
1. Leblanc R. Familial adenomatous polyposis and benign intracranial tumors: a new variant of Gardner’s syndrome. Can J Neurol Sci. 2000;27(4):341-6.
2. McLaughlin MR, Gollin SM, Lese CM, Albright AL. Medulloblastoma and glioblastoma multiforme in a patient with Turcot syndrome: a case report. Surg Neurol. 1998;49:295-301. [ Links ]
3. Sunahara M, Nakagawara A. Turcot syndrome. Nihon Rinsho. 2000;58(7):1484-9. [ Links ]
4. Turcot J, Després JP, ST Pierre F. Malignant tumors of the central nervous system associated with familial polyposis of the colon: report of two cases. Dis Colon Rectum. 1959;2:465-68 [ Links ]
5. Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, et al. The molecular basis of Turcot’s Syndrome. N Engl J Med 1995;332(13):839-47.
6. Sahnane N, Bernasconi B, Carnevali I, Furlan D, Viel A, Fausto Sessa F, et al. Disruption of the APC gene by t(5;7) translocation in a Turcot family. Cancer Genet. 2016;209(3):107-11. [ Links ]
7. Ozerov S, Zakharov I, Talypov S. Turcot syndrome. A rare case and literature review. Zh Vopr Neirokhir Im N N Burdenko. 2013;77(3):49-53. [ Links ]
8. Castro M, Sullcahuaman Y, Barreda F, Taxa L. Síndromes hereditarios de predisposición al cáncer colorrectal identificados en el Instituto Nacional de Enfermedades Neoplásicas, Lima, Perú. Rev Gastrenterol Peru. 2014;34(2):107-14- [ Links ]
9. Shun Chang M, Ye Shuai H, Jun Y, Ziao Na Z. Turcot’s syndrome associated with intestinal non-Hodgkin’s lymphoma: Case report and review of literature. Clin Neurol Neurosurg. 2013;115(2):117-20.
10. Kropilak M, Jagelman DG, Fazio VW, Lavery IL, McGannon E. Brain tumors in familial adenomatous polyposis. Dis Colon Rectum. 1989;32(9):778-82. [ Links ]
11. Skomorowski M, Taxier M, Wise W. Turcot syndrome type 2: Medulloblastoma with multiple colorectal adenomas. Clin Gastroenterol Hepatol. 2012;10(10):A24. [ Links ]
12. Buecher B. Colorectal adenomatous polyposis syndromes: Genetic determinism, clinical presentation and recommendations for care. Bull Cancer. 2016;103(2):199-209. [ Links ]
13. Vallejo D, Garnica D, Bonilla R, Olaya N. First case report of Turcot syndrome type 1 in Colombia. Case Rep Oncol Med. 2012:356384. doi:10.1155/2012/356384. [ Links ]
Correspondencia:
Carlos Barreda Costa Clínica Ricardo Palma. Av Javier Prado Este 1060, San Isidro, Lima, Perú.
E-mail: cbarreda@gastrobasamea.com

