











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Las glucogenosis o enfermedades del almacenamiento de glucógeno, abarcan un rango de enfermedades que se caracterizan por el almacenamiento o utilización anormal del glucógeno.
Más de 12 tipos de glucogenosis han sido identificadas, las cuales son clasificadas de acuerdo a su deficiencia enzimática. En general, las glucogenosis afectan primariamente el hígado y el músculo, pero pueden también afectar los riñones o intestino 1.
La hipoglicemia recurrente y hepatomegalia 2 son las características cardinales de las glucogenosis que afectan el hígado, mientras que rabdomiólisis, dolor muscular o cardiomiopatía son signos de glucogenosis afectando los músculos.
Es importante identificar el tipo de glucogenosis ya que cada uno tiene una historia natural, tratamiento y complicaciones diferentes.
A continuación, presentamos el caso de un adolescente quien desde la infancia presentaba hepatomegalia, la cual fue diagnosticada erróneamente como "fisiológica", llevando a un diagnóstico y tratamiento tardío de la glucogenosis hepática, desencadenando retraso en su crecimiento y desarrollo.
CASO CLÍNICO
Paciente varón, natural de Trujillo, producto de primera gestación, embarazo controlado, sin complicaciones. Madre y padre aparentemente sanos, sin consanguinidad entre ellos.
Peso al nacer 3000 g, talla de 50 cm, no hipoglicemia al nacimiento. A los 4 años de edad fue hospitalizado por presentar hepatomegalia y retraso en el crecimiento, siendo dado de alta con diagnóstico de "hepatomegalia fisiológica".
Referido al consultorio de Gastroenterología del Hospital de Alta Complejidad "Virgen de la Puerta" a la edad de 15 años por presentar hepatomegalia e hipertransaminasemia; así como dolor abdominal difuso en hipocondrio derecho de un año de evolución.
Al examen físico se encontró: peso: 41,5 Kg, talla: 147 cm (valores por debajo del percentil 10 para su edad), abdomen globuloso, presencia de circulación colateral, spam hepático de aproximadamente 12 cm.
Exámenes auxiliares: aspartato aminotransferasa: 382 U/L (0-34 U/L), alanina aminotransferasa: 189 U/L (10-49 U/L), bilirrubina total: 2,6 mg/dl (hasta 1 mg/ dl), bilirrubina indirecta: 1,8 mg/dl (hasta 0,8 mg/dl), albúmina: 4,4 g/dl (3,5-5 g/dl), colesterol: 174 mg/dl (135-200 mg/dl), triglicéridos: 182 mg/dl (45-150 mg/ dl), lipoproteína de alta densidad: 14 mg/dl (35-55 mg/ dl), lipoproteína de baja densidad: 123 mg/dl (menor de 130 mg/dl). Glucemia basal 89 mg/dl (70-110 mg/ dl), dosaje de insulina 47,34 (70 a 110 UI), factor de crecimiento insulinico-1: 45 (116-200 ng/ml).
Se descartaron las principales causas de hipertransaminasemia: hepatitis viral B y C, toxoplasmosis, citomegalovirus y marcadores de enfermedad hepática autoinmune negativos. Perfil de hierro: saturación de transferrina, ferritina e Inmunoglobulinas dentro de rangos de normalidad.
Cortisol en ayunas y perfil tiroideo sin alteraciones. Proteinuria al azar 29,5 mg/l, creatinina en orina al azar 49,46 mg/24h, cetonas en orina negativo, creatinina en sangre: 0,4 mg/dl (0,8-1,4 mg/dl) ácido úrico: 3,5 mg/ dl (3,5-7 mg/dl), creatinquinasa: 1517 (24-195) ácido láctico 1,3 mmol/l (menor de 2 mmol/l).
Ecografía doppler portal describió hígado de 133 mm de longitud, ecogenicidad incrementada, parénquima homogéneo, no presencia de lesiones focales, no dilatación de vía biliar intrahepática. Vena porta de 8,7 mm de diámetro, flujo sin alteraciones.
Ecocardiograma y fondo de ojo no patológicos.
Endoscopía digestiva alta sin hallazgos significativos.
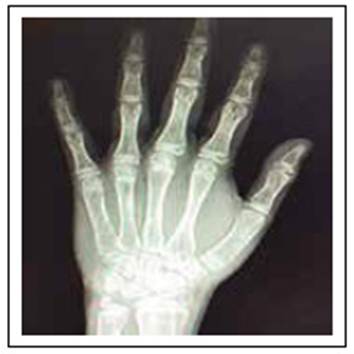
Radiografía de mano izquierda: edad ósea 12,7 años (ver Figura 1).
Por evidencia de hipertransaminasemia crónica sin causa determinada, teniendo sospecha de enfermedad hepática por depósito, se decidió realizar una biopsia hepática quirúrgica.
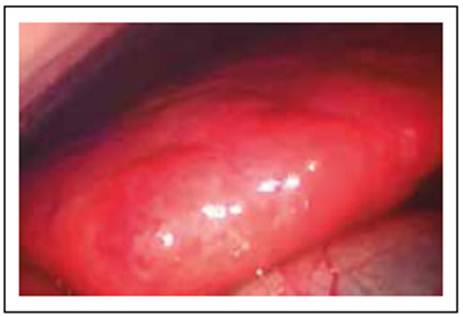
Durante la cirugía se observó hepatomegalia que abarca todo el hemiabdomen superior, superficie hepática micro nodular, color pardo, de bordes romos (ver Figura 2). A nivel del segmento 3 se realizó biopsia en cuña.
Los hallazgos histológicos revelaron aumento del glucógeno intrahepatocitario, hallazgos en relación a glucogenosis hepática (ver Figuras 3 y 4).
DISCUSIÓN
Las glucogenosis son un grupo de enfermedades hereditarias, caracterizadas por la alteración en el depósito del glucógeno, a causa de una deficiencia genética de la actividad de alguna de las enzimas que degradan o sintetizan al glucógeno, siendo los dos tejidos más afectados el músculo y/o el hígado.
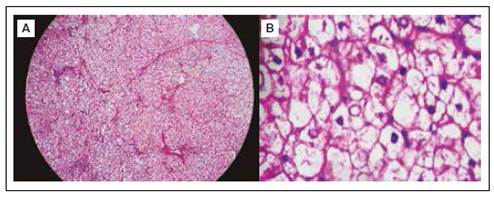
Figura 3 Histología de biopsia hepática. (A) Tinción: hematoxilina-eosina. Hepatocitos con citoplasma claro, patrón de mosaico debido a la compresión de los sinusoides. (B) Tinción: hematoxilina-eosina. Membrana citoplasmática de hepatocitos engrosada, citoplasma expandido claro por el depósito de glucógeno, núcleo glucogenado.
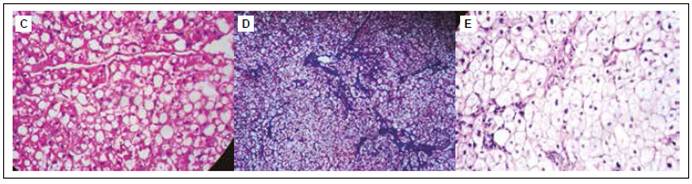
Figura 4 Histología de biopsia hepática. (C) Tinción: hematoxilina-eosina. Esteatosis macrovesicular a gota grande en zona acinar 1. (D) Tinción: Tricrómica de Masson. Algunos espacios porta con ampliación fibrosa, fibrosis perisinusoidal y pericelular. (E) Tinción: PAS-Diastasa. Hepatocitos mostrando la digestión de glucógeno (+).
Los diferentes tipos de glucogenosis son clasificados de acuerdo al déficit enzimático y los órganos afectados 3.
Las manifestaciones clínicas son la expresión de la dificultad para movilizar los depósitos de glucógeno en los tejidos afectados. Cuando el hígado es afectado, se produce hepatomegalia, alteración en la regulación de la glicemia y retardo en el crecimiento.
Si bien el diagnóstico se basa en la clínica, las determinaciones bioquímicas, como glicemia, cuerpos cetónicos, perfil lipídico, así como el test de tolerancia a glucosa aportan datos para el diagnóstico diferencial entre los tipos de glucogenosis.
La biopsia hepática confirma el aumento de glucógeno intrahepatocitario, además de identificar el grado de fibrosis.
En nuestro caso, el paciente presentó hepatomegalia, retardo en el crecimiento, así como elevación de las transaminasas, creatinfosfoquinasa y un discreto aumento de los triglicéridos. No presentó hipoglicemia ni cetonuria. El ácido láctico y ácido úrico se encontraban dentro de rangos normales
No presentó compromiso, renal, cardiaco o muscular; por lo que podemos inferir que lo más probable es que se trate de una glucogenosis tipo VI (enfermedad de Hers) o tipo IX (Tabla 1).
La glucogenosis tipo IX resulta de la disfunción de la enzima fosforilasa quinasa, que está compuesta por 4 subunidades. Existen diferentes subtipos dependiendo de la subunidad afectada 4.
La presentación clínica es muy heterogénea y variable, puede ir desde hepatomegalia e hipertransaminasemia, hasta hipoglicemia y cirrosis 5.
La enfermedad de Hers se caracteriza por el déficit de la fosforilasa hepática E (cromosoma 14q21-q22) 6, siendo la forma más rara y probablemente la más subdiagnosticada 7, ya que presenta un cuadro clínico leve.
Los pacientes presentan en la infancia o niñez distensión abdominal, hepatomegalia, retardo del crecimiento. Si presentan hipoglicemia, esta es leve y se manifiesta después ayuno prolongado 8.
Individuos con glucogenosis tipo IX exhiben fenotipo muy similar a la glucogenosis tipo VI 9, excepto que la herencia de la glucogenosis IX es ligada al X y la segunda es autosómica recesiva.
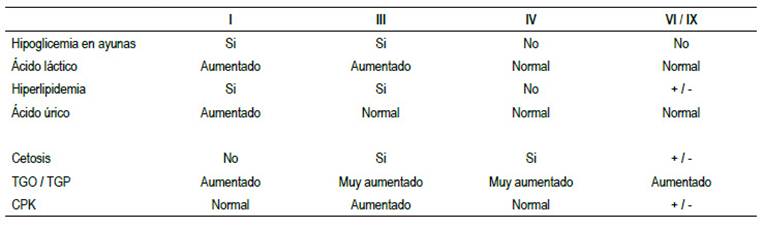
El diagnóstico diferencial de estos dos tipos se realiza por medio de genética molecular 8 examen que no se pudo realizar por no tenerlo disponible.
La biopsia hepática muestra hepatocitos en patrón de mosaico, y distensión irregular de los hepatocitos debido al depósito de glicógeno, como fue visto en el caso de nuestro paciente 5.
En el presente caso, existió retraso en el diagnóstico de la enfermedad, ocasionando un retardo en el desarrollo del paciente, como lo muestra en las medidas antropométricas que revelan talla baja para la edad y se correlaciona con la radiografía de mano izquierda, la que reporta edad ósea de 12,7 años.
Generalmente, el curso clínico es benigno y las alteraciones clínicas y bioquímicas, así como la hepatomegalia disminuyen con la edad, tanto así, que muchos adultos permanecen asintomáticos 10-12.
El principal objetivo en la terapia de la glucogenosis VI y IX es prevenir las manifestaciones primarias (hipoglicemia, cetosis y hepatomegalia) y complicaciones secundarias (pubertad retardad, baja estatura y cirrosis) 13.
Esto se logra al mantener niveles óptimos de glicemia simulando las demandas endógenas. Se usa una dieta alta en carbohidratos y fraccionada 13.
El paciente actualmente está en tratamiento dietético con buena respuesta, ha disminuido la hepatomegalia, así como discretamente los valores de transaminasas, en radiografía control a los 2 años de tratamiento, la edad ósea es de 13 años y 6 meses.
En conclusión, el caso descrito sugiere que, ante la presencia de hepatomegalia en infantes o adolescentes, se debe considerar la posibilidad diagnóstica de enfermedad por depósito. El realizar un diagnóstico oportuno conllevara a minimizar las complicaciones, como el retraso en el crecimiento y desarrollo de este adolescente con diagnóstico tardío de glucogenosis hepática.

