











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Las porfirias son trastornos metabólicos de baja incidencia. La más común a nivel global es la porfiria cutánea tarda (PCT), seguida de la protoporfiria eritropoyética (EPP) 1. En Colombia la EPP tiene incidencia de 0,2 y prevalencia de 15,4 por cada millón de habitantes 2. La porfiria aguda intermitente (PAI) es la más frecuente de las porfirias agudas, tiene herencia autosómica dominante y una tasa estimada de 0,5-1: 75 000 - 100 000 en algunos países europeos, en América Latina la prevalencia en la población Argentina es de 1: 125 000 3-5. La prevalencia exacta se desconoce debido a penetrancia incompleta y baja sospecha diagnostica.
Las porfirias hepáticas agudas son PAI, porfiria del ácido delta aminolevulinico deshidratasa (ALAD), coproporfiria hereditaria (CPH) y porfiria variegata (PV) afectan aproximadamente a 20 000 personas, generalmente a mujeres en la tercera década de la vida, aunque es rara presentación previa a pubertad y después de menopausia, la ALAD la cual es más común en hombres y suele manifestarse antes de la pubertad o edad adulta se caracterizan por presentes síntomas agudos y crónicos por su compromiso sobre el sistema nervioso, el síntoma más común es dolor abdominal neuropático 3,5-7.
Las porfirias eritropoyeticas son la EPP y la porfiria eritropoyética congénita (CEP). También se pueden clasificar según presencia o ausencia de lesiones ampollosas, las ampollosas son la PCT, CEP, PV y CPH, las no ampollosas EPP y la protoporfiria ligadas al cromosoma X (XLP) y protoporfirinas congénitas LPX que son extremadamente raras 3,6,7; de las cuales ninguna presenta manifestaciones neurológicas. A continuación, se presenta 3 casos clínicos de interés por su presentación clínica y complicaciones potencialmente prevenibles al realizar intervención temprana.
CASOS CLÍNICOS
Caso 1:
Se trata de una paciente de 32 años, quien ingresó al servicio de urgencias por presencia de dolor abdominal generalizado de 4 días de evolución, a los 2 días de inicio de los síntomas presentó sensación de parestesias en miembros inferiores, progresó a miembros superiores asociado a debilidad distal de 4 extremidades y disartria. En el seguimiento intrahospitalario, presentó deterioro del patrón respiratorio sin requerimiento de ventilación mecánica invasiva. Una semana después presentó coluria. Se consideró sospecha de porfiria aguda intermitente y se solicitó porfobilinogeno en 24 horas, que es positivo. Se documentó una polineuropatía axonal motora pura en las neuroconducciones. Se indicó hemina durante 4 días con mejoría clínica.
Caso 2:
Se trata de una paciente de 21 años, quien ingresó al servicio de urgencias por dolor abdominal agudo generalizado, presencia de delirium y crisis epiléptica, al ingreso presentó orina colúrica. Se sospechó porfiria aguda intermitente, se solicitó porfobilinógeno con resultado positivo. Se indicó hemina con mejoría de los síntomas.
En una hospitalización posterior, presentó nuevo episodio de dolor abdominal con deterioro del patrón respiratorio con posterior progresión a falla ventilatoria por cuadriparesia flácida. Requirió traqueostomía y gastrostomía por el trastorno deglutorio. Presentó atrofia muscular, cuadriparesia y dolor neuropático con presencia de disestesias y parestesias de predominio distal.
En el seguimiento se logró oclusión de traqueostomía y gastrostomía, mejoría de fuerza en 4 extremidades.
Caso 3:
Mujer de 22 años con dolor abdominal severo y estreñimiento de 4 días. Se llevó a cirugía por sospecha de vólvulo sin documentarse hallazgos intraoperatorios anormales. Cursó además con 7 días de debilidad progresiva proximal en miembros inferiores y superiores. Posteriormente, presentó crisis epiléptica e hiponatremia. Durante evolución cursó con disautonomía y coluria. Se documentó polineuropatía axonal sensitivo motora aguda.
Se confirmó porfobilinógeno en orina, se inicia hemina. Su evolución fue tórpida progresa a falla ventilatoria requirente soportes, complicaciones derivadas de estancia hospitalaria finalmente paciente fallece posterior a 3 paros cardiacos.
Fisiopatología
El grupo hemo se produce principalmente en células eritropoyéticas para síntesis de citocromos, hemoproteínas, y hemoglobina en médula ósea. Ocho enzimas componen esta ruta metabólica. El primer paso es en la enzima delta-aminolevulinato sintasa (ALAS), que convierte glicina y Succinil- CoA con la ayuda del cofactor piridoxal-5-fosfato (derivado de la vitamina B6) en ácido delta-aminolevulínico (ALA) exclusivo para la síntesis de hemo 1,2,7.
ALAS tiene dos presentaciones: ALAS1 y ALAS2. La primera se encuentra ubicada en el cromosoma 3 y hace referencia en este caso a las células hepáticas, mientras que la segunda se encuentra ubicada en el cromosoma X y se produce solo en eritroblastos de la médula ósea. ALAS1 es la enzima que limita la velocidad en que se produce hemo y se controla por retroalimentación negativa del hemo libre no comprometido con la síntesis de hemoproteínas, mientras ALAS2 es limitada por la disponibilidad de hierro y no se ve inhibida por el hemo 2,8,9. En ALAS1 no se ha visto una mutación específica, pero sí se ha visto que la sobreexpresión de este es importante en las porfirias hepáticas agudas. Por otro lado, mutaciones en ALAS2 han causado anemia sideroblástica ligada al sexo o protoporifira ligada al cromosoma X 3.
Continuando, la ALA desintegrasa (ALAD) convierte el ALA en porfobilinógeno (PBG). Hasta ahora, tanto ALA como PBG se conocen comúnmente con los precursores de porfirina los cuales son potencialmente neurotóxicos. Luego la PBG desaminasa (PBGD) o también conocida como hidroximetilbilano sintasa (HMBS) convierte el PBG en hidroximetilbilano (HMB) 1,2,8. Posteriormente el HMB, que es un tetrapirrol lineal, se reorganiza y se cicla con una inversión de un pirrol para formar la primera porfirina asimétrica conocida como uroporfirinógeno III gracias a la uroporfirinógeno sintasa (UROS) 3. El resto de HMB que haya sobrado se cicla en forma no enzimática y se forma el uroporfirinógeno I simétrico 3. El siguiente paso es que la uroporfirinógeno descarboxilasa (UROD) convierte el uroporfirinógeno I o III en coproporfirinógeno I o III. Posteriormente, la coproporfirinógeno oxidasa (CPOX) convierte exclusivamente el coproporfirinógeno III en protoporfirinógeno IX. En el antepenúltimo paso, la protoporfirinógeno oxidasa (PPOX) oxida en protoporfirinógeno IX eliminando seis protones y forma la protoporfirina IX que es el único intermedio de porfirina oxidado en la vía. Finalmente, la ferroquelatasa (FECH) inserta el hierro en la protoporfirina IX para formar el hemo. Las enzimas ALAS, CPOX, PPOX y FECH son mitocondriales, y las cuatro intermediarias ALAS, PBGD, UROS y UROD son citoplasmáticas. La anormalidad de una enzima por déficit o sobreexpresión ocasiona los distintos tipos de porfirias, tienen patrón de transmisión genética mendeliana y no mitocondrial 3.
A pesar de que las porfirias que presentan disfunción neurológica tengan déficit en diferentes enzimas (PAI en PBGD, porfiria del ácido delta aminolevulinato en ALAD, VP en PPOX y CPH en CPOX) hay algo en común que tienen y es la sobreestimulación de ALAS1. Realmente se desconoce el mecanismo exacto de la disfunción neurológica que se da, pero la hipótesis principal es que ALA y otros metabolitos sobre producidos por el hígado son neurotóxicos y pueden generar disfunción metabólica celular generando un deterioro de energía intracelular 2,7,9,10. Se ha visto que ALA puede atravesar la barrera hematoencefálica y generar efectos tóxicos directos en el cerebro tras presentar un proceso de autooxidación que genera radicales libres que conducen a peroxidación lipídica, además de generar daño oxidativo en las mitocondrias del hígado 10 11. El estrés oxidativo se ha propuesto como un factor importante en los desórdenes oxidativos que se ha respaldado por experimentos in vitro donde ALA induce inhibición de la formación de mielina por los oligodendrocitos 11.
DISCUSIÓN
Las crisis de porfirias aguda son poco frecuentes y son ocasionadas por acumulación de precursores de porfirinas como PBG y ALA. El 5% de las crisis se presentan en menores de 14 años y se deben considerar como diagnostico diferencial en dolor abdominal de etiología desconocida en infancia tardía y adolescencia. Tienen mortalidad de 20 a 25% en los primeros 5 años después del primer evento 1,11,12.
El compromiso gastrointestinal, se caracteriza por dolor abdominal (85-95% de pacientes) de intensidad severa, constante mal localizado que, con frecuencia, como se describió, obliga a la realización de cirugía abdominal sin documentarse hallazgos anormales. Además, hay disautonomía dada por taquicardia (en el 80%) o diaforesis. Entre los síntomas neuropsiquiátricos el más temprano es insomnio, pero se ha descrito también agitación, ansiedad, alucinaciones, apatía, depresión, alteración del estado de conciencia desde somnolencia hasta coma. Se puede presentar parálisis bulbar, compromiso en pares craneales, neuropatías periféricas, en casos severos parálisis flácida con progresión a tetraparesia y disfunción de músculos respiratorios con riesgo de falla ventilatoria y muerte. La tetraparesia es aguda y secundaria a una polineuropatía axonal. Clásicamente se describe un compromiso motor puro, pero no es infrecuente encontrar compromiso sensitivo-motor como el encontrado en las pacientes. Esta polineuropatía simula un síndrome de Guillain-Barré y es potencialmente reversible con rehabilitación, sin embargo, en ocasiones el daño es permanente. Se ha descrito también cambios de color en orina posterior a exposición a luz solar 1,11,12.
Se deben identificar presencia de factores predisponentes para las crisis de porfirias, entre ellos el uso de algunos medicamentos no seguros (Tabla 1) que ocasionan inducción de ALAS1 y enzimas del citocromo P450 13.
Tabla 1 Seguridad de medicamentos en pacientes con porfiria.
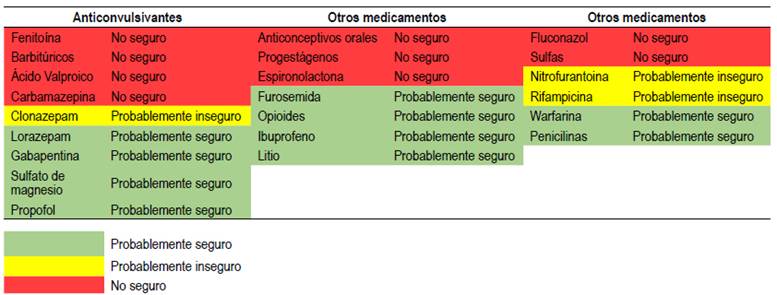
Adaptado de Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. N Engl J Med. 2017;377(9):862-72.
Las crisis neuroviscerales se presentan en PAI, HCP, VP, ADP las cuales tienen características resumidas en la (Tabla 2). Estas tienen dos variantes, una manifiesta y otra latente (portador con potencial riesgo de desarrollo de enfermedad). El diagnóstico de la manifiesta se basa en medición de actividad deficiente de PBGD en eritrocitos y/o identificando una mutación de PBGD / HMBS en un individuo con síntomas y hallazgos bioquímicos compatibles con PAI (síntomas neuroviscerales y PBG urinario sustancialmente elevado, con poca o sin elevación de las porfirinas plasmáticas y fecales). La prueba de mutación del gen PBGD/HBMS es la más confiable y se detecta en el 95% de pacientes con AIP, aproximadamente el 90% tendrá una actividad deficiente de PBGD eritrocitaria 1,9,14,15.
Tabla 2 Clasificación de porfirias en formas cutáneas y no cutáneas.
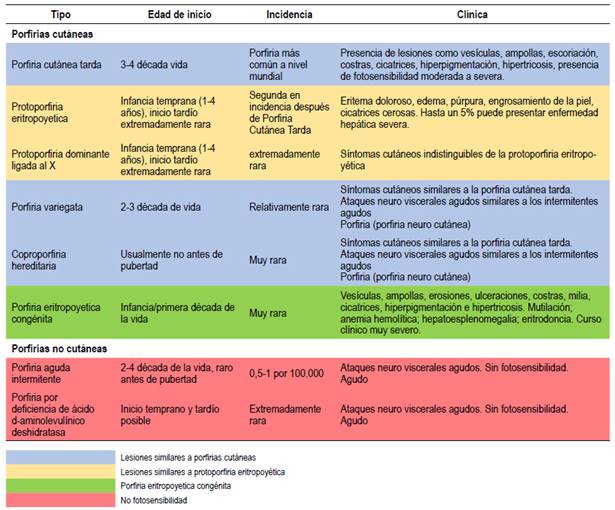
Adaptado de Livideanu CB et al. Late-onset X-linked dominant protoporphyria: An etiology of photosensitivity in the elderly. J Invest Dermatol. 2013;133(6):1688-90
Respecto a la PAI latente se identifica mutación del gen que afecta la actividad de PBGD en un individuo sin síntomas de AIP. También se utiliza el término para individuos con ausencia de síntomas durante años con o sin elevación de PBG. La PBG en orina es la prueba de detección de primera línea más importante cuando se sospecha AHP 1.
-Si los resultados son positivos (p. Ej., Nivel de PBG> 10 mg por g de creatinina [o> 10 mg/L]), el tratamiento con hemina puede (y normalmente debe) iniciarse sin demora si las manifestaciones clínicas son graves
-Si los resultados son negativos (p. Ej., Nivel de PBG <5 mg por g de creatinina [o <5 mg/L]), es apropiado realizar pruebas para otras condiciones en el diagnóstico diferencial 1.
Se deben obtener muestras adicionales para determinar el tipo de AHP antes de iniciar el tratamiento. Obtenemos las siguientes muestras:
-Porfirinas en orina
-Porfirinas plasmáticas
-Porfirinas fecales
* No se necesitan recolecciones extendidas (24 horas). 1
El manejo para las porfirias agudas depende de la severidad del cuadro clínico 12). Se deben identificar los potenciales desencadenantes del evento, entre ellos fármacos no seguros en porfirias. Al examen físico se debe evaluar la presencia de cifras tensionales elevadas o taquicardia. En caso de signos de dificultad respiratoria se deberá trasladar de forma temprana al paciente a unidad de cuidados intensivos por riesgo de falla ventilatoria y requerimiento de ventilación mecánica 3,11,12,16.
La hiponatremia es el trastorno hidroelectrolítico más común. Es secundario a SIADH (síndrome de secreción inadecuada de ADH), niveles moderados a severos es indicación de inicio de Hemina. Se debe evaluar la función renal y niveles de creatina-fosfocinasa (CPK) debido a que lesión renal aguda y rabdomiólisis son complicaciones infrecuentes de porfiria, pero potencialmente mortales 12.
Una crisis se considera moderada a severa si presenta encefalopatía, debilidad muscular, alteración respiratoria, hiponatremia moderada a severa o dolor abdominal que requiera hospitalización y uso de opioides de alta intensidad para manejo sintomático 12.
En crisis moderadas a severas está indicado el uso de Hemina (Heme argintato), actúa suprimiendo la hiperfunción del primer paso enzimático ALAS1 por suplencia del heme. La supresión de esta enzima ocasiona reducción en la síntesis de los precursores tóxicos ALA y porfobilinogeno. Se administran 3-4 mg/kg disueltos en 100 ml de solución salina al 0,9% por vía intravenosa central en dosis única al día con infusión en 30 minutos. Su dosis máxima es 250 mg/día durante 4 días 12. Se debe proteger la dilución de la luz solar con cobertura opaca o de aluminio. La hemina se puede administrar durante el embarazo 12.
La dextrosa está indicada en crisis agudas leves, al reducir la excreción de precursores de porfirina por efecto represivo en inducción de ALAS1, tiene menor efecto respecto a hemina. Se puede utilizar dextrosa al 10% intravenosa con 500 ml cada 6 horas, total 200 gramos 12.
El manejo del dolor se basa en opioides de baja y alta intensidad los cuales son medicamentos seguros y efectivos en porfirias. Se debe evitar la dipirona por su potencial porforinogénico. Ondansetrón y clorpromazina se prefieren sobre la metoclopramida para el manejo de náuseas y emesis. Las crisis epilépticas son secundarias a disfunción del sistema nervioso central o a hiponatremia severa. Se pueden utilizar de forma segura y efectiva levetiracetam y benzodiacepinas. Se deben evitar barbitúricos y carbamazepina por su potencial porfirinogénico. La hipertensión y taquicardia se beben manejar con betabloqueadores como labetalol, atenolol y propranolol. Es importante una vigilancia estricta debido al riesgo de disautonomía, hipotensión, bradicardia y deshidratación 12.
En caso de infección se debe tratar de forma temprana con cubrimiento antibiótico según el foco sospechado, se deben evitar sulfonamidas por su potencial porforinogénico 12.
En conclusión, es importante considerar el diagnóstico de porfiria aguda en pacientes jóvenes con dolor abdominal, difuso, inespecífico y severo, sobre todo si han tenido antecedentes de enfermedad psiquiátrica (ansiedad, psicosis o depresión). Así mismo, como se documentó en los casos descritos, los pacientes pueden cursar con dolor abdominal y una polineuropatía aguda similar a un síndrome de Guillain-Barré de características axonales y de predominio motor.
Es fundamental reconocer las manifestaciones clínicas tempranamente para prevenir las complicaciones y potenciales secuelas.

