










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. v.64 n.1 Lima ene./mar. 2003
Disfunción endotelial en la preeclampsia
Endothelial dysfunction in pre-eclampsia
José Pacheco Romero
Profesor Principal de Obstetricia y Ginecología, Facultad de Medicina San Fernando, UNMSM.
Jefe de la Unidad de Reproducción Humana, Hospital Nacional Edgardo Rebagliati Martins, EsSalud.
Resumen
Desconocemos aún la etiología de la preeclampsia, pero ahora sabemos que no es sólo una hipertensión inducida por el embarazo, sino que existe interacción entre una perfusión placentaria disminuida y la alteración en la función endotelial materna, probablemente por razones inmunológicas de rechazo parcial a la placentación normal. La contribución materna es de factores que anteceden al embarazo, influenciados por las adaptaciones metabólicas usuales. No existe un gen único que pueda explicar la preeclampsia, pero conocer la predisposición materna permite prevenir la preeclampsia en un grupo de mujeres.
Palabras clave: Preeclampsia; eclampsia; endotelio; perfusión; placentación.
Summary
We still do not know the etiology of pre-eclampsia. We do know now that it is not only a pregnancy induced hypertension, but an interaction between decreased placental perfusion and deep maternal endothelial function alteration, probably due to immune reasons favouring partial alteration of normal implantation. Maternal contribution is of factors that predate pregnancy influenced by the usual metabolic adaptations. There is no single gene to explain preeclampsia, but looking for maternal predisposition may prevent pre-eclampsia in a group of women.
Key words: Pre-eclampsia; eclampsia; endothelium; perfusion; placentation.
INTRODUCCIÓN
Hemos ingresado al Siglo XXI y seguimos sin conocer la etiología de la preeclampsia-eclampsia. Por los nuevos conocimientos de la fisiopatología de la enfermedad, la denominación de hipertensión inducida por el embarazo –utilizada en la última década- no se ajusta a los hallazgos clínicos de la enfermedad. Así, mientras la hipertensión es un signo importante del proceso, ella es consecuencia de la enfermedad y no la causa, y parecería tener el papel de compensar la disminución del flujo sanguíneo materno fetal (1). Además, a pesar que la proteinuria no siempre ocurre y el edema se presenta en muchas mujeres sin la enfermedad, se ha preferido regresar al término de preeclampsia-eclampsia, por la presencia de estos signos en la enfermedad, aunque todos ellos pueden estar ausentes. En algunas mujeres, la compensación a las variaciones fisiológicas del embarazo se resquebraja y ocurre la enfermedad multisistémica severa (2) que los ginecoobstetras, nefrólogos, inmunólogos, cardiólogos –entre otros- conocemos como preeclampsia.
La morbimortalidad materna asociada a la preeclampsia es por hemorragia intracerebral, eclampsia o por disfunción de órgano terminal, siendo secuelas de la enfermedad la hipertensión secundaria persistente, morbilidad neurológica y alteración renal.
La morbimortalidad perinatal es reflejo de la restricción del crecimiento intrauterino (RCIU) y de prematuridad yatrogénica (25% de bebes con peso bajo nace con peso por debajo de 1500 g) (3), debida al deterioro materno o de la condición fetal. Es así, que las unidades de cuidados intensivos neonatales hoy están enriqueciendo su experiencia en la atención de bebes de peso muy bajo, tanto pretérmino como con restricción en su crecimiento, gracias a –diríamos mejor a causa de- la mayor frecuencia de preeclampsia-eclampsia en nuestros hospitales.
El conocimiento de la preeclampsia y eclampsia es importante en el Perú por la alta morbimortalidad materna y perinatal que ocasiona. Para el año 2002, la incidencia de hipertensión en el embarazo en Lima y Trujillo alcanzaba hasta 15% (4,5), la eclampsia 8% (6,7) y el síndrome HELLP 3,7%o (8) (Tabla 1). Es decir, parece que la enfermedad tiene cada vez mayor prevalencia, especialmente en los hospitales de tercer y cuarto nivel, representando en EsSalud la primera causa de muerte materna y causa importante de mortalidad perinatal.

Esta alta prevalencia de preeclampsia-eclampsia en el Perú, que al momento pasaría el 15% en varias instituciones hospitalarias en el país, nos obliga a conocer con mayor profundidad su fisiopatología.
EL ENDOTELIO
El endotelio ha sido visto por largo tiempo como una membrana inerte, semipermeable, existente entre la sangre y la pared del vaso. Hoy día dicho concepto ha cambiado totalmente y se observa al endotelio como un importante órgano endocrino, grande y muy activo, responsable de un número de funciones fisiológicas vitales (15).
Se encuentra mensajero ARN (ARNm) y proteína del receptor de hormona gonadotropina coriónica/hormona luteinizante (hCG/LH) en las capas del músculo liso endotelial y vascular de las arterias del útero. La administración in vivo de hCG disminuye la resistencia de flujo en el útero e, in vitro, aumentan los eicosanoides vasodilatadores en la pared vascular. En la amenaza de aborto, se observa que la tasa de pacientes que llegan al segundo trimestre aumenta y los pretérminos y bebes con restricción del crecimiento intrauterino (RCIU) disminuyen cuando se administra hCG en el primer trimestre (16).
El tono vascular depende del estímulo del nervio neurogénico periarterial y de la secreción de sustancias endoteliales. La causa bioquímica más evidente del proceso contracción/relajación del músculo liso vascular reside en la variación en la concentración de Ca2+ citosólico. Así, el calcio libre intracelular es el mayor determinante del tono vascular. La disfunción de la célula endotelial se acompaña por disminución en la producción y/o secreción de óxido nítrico (NO) y aumento de los factores contráctiles. Ello induce a la movilización de Ca2+ de los depósitos extra- e intracelulares. La contracción del músculo liso ante la hipoxia es mediada por acúmulo de Ca2+ intracelular. Es válido recordar que la preeclampsia se asocia en muchas gestantes con ingesta dietética baja de calcio, que aparentemente mejora con la administración de suplementos de calcio (17).
El endotelio intacto tiene propiedades anticoagulantes. Mientras tanto, el endotelio dañado activa las células endoteliales y aumenta la sensibilidad a los agentes vasopresores, promoviendo la coagulación (18).
Enseguida revisaré las sustancias producidas por el endotelio y cómo se alteran en la preeclampsia la contractilidad vascular, la coagulación, activación del neutrófilo, alteración de los lípidos, entre otros, para luego detenerme en lo que se conoce sobre la placentación normal, incluyendo las modificaciones en endotelio y vasos uterinos, y las variaciones en la preeclampsia.
SUSTANCIAS ENDOTELIALES
El endotelio influye en el tono vascular produciendo sustancias relajantes o vasodilatadoras –prostaciclina (PGI2), óxido nítrico (NO, derivado de l-arginina) y factor hiperpolarizante derivado del endotelio (probable metabolito del ácido araquidónico)- y vasoconstrictoras –endotelina, tromboxano (TxA2) y aniones superóxidos-. También el tono vascular está muy ligado al vasodilatador péptido natriurético atrial (ANP) y al sistema renina-angiotensina. Además, el endotelio expresa ectopeptidasas, que convierten la angiotensina I en angiotensina II, inactivan la bradiquinina y producen endotelina activa de la endotelina grande (18).
La prostaciclina y el óxido nítrico pueden inhibir la activación de las plaquetas, neutrófilos y de sustancias, tales como el activador del plasminógeno tisular (TPA), que previenen o limitan la coagulación y el daño vascular. Los glicosaminoglicanos de la membrana plasmática de la célula endotelial son ricos en sulfatos de heparina y así se unen a la antitrombina, aumentando su afinidad por la trombina, permitiendo su rápido aclaramiento.
Óxido nítrico
En el embarazo, la presión arterial disminuye en el primer trimestre, hace nadir en la mitad del embarazo y aumenta luego lentamente; el output cardíaco aumenta 40% y disminuye la reactividad al estímulo presor. La disminución de la presión arterial es por disminución de la resistencia vascular sistémica. En la segunda mitad del embarazo aumenta la resistencia vascular. El óxido nítrico (NO) contribuye a modular el tono en el árbol vascular de la villosidad placentaria. Recordemos que la circulación fetoplacentaria no tiene inervación (19,20).
Es decir, el óxido nítrico tiene papel importante en el control de la presión arterial sistémica. La sintasa del óxido nítrico endotelial (e-NOS) genera NO continuamente, que se difunde en el músculo subyacente, aumenta la producción de cGMP y así media la vasodilatación. Es inhibidor de la activación de las plaquetas y neutrófilos y, si no se forma, activará los neutrófilos, habrá vasoconstricción, adhesión y agregación plaquetaria y liberación de sustancias vasoconstrictoras.
Tratando de determinar la genética de la preeclampsia, se ha observado que las mujeres con cambios en un gen que parte partículas de grasa tienen 4 veces el riesgo de desarrollar preeclampsia (21). El gen de la sintasa del óxido nítrico de la célula endotelial humana (eNOS) es candidata de la susceptibilidad a la preeclampsia/eclampsia. Es posible que el locus resida en la región 7q36 (22).
Desde que el sincitiotrofoblasto delinea la superficie placentaria y está en contacto directo con la circulación materna, el NO producido en el sincitiotrofoblasto puede prevenir la adhesión de plaquetas y leucocitos en el espacio intervilloso (18).
Prostaciclina
La prostaciclina (PgI2) derivada del endotelio es un potente vasodilatador, antiagregante plaquetario y estimula la secreción de renina. En contraste, el tromboxano A2 (TxA2), liberado por las plaquetas y metabolizado de endoperóxidos por la sintasa de tromboxano, es un potente vasoconstrictor y agregante plaquetario. Es decir, estos derivados eicosanoides tienen efectos contrarios en la trombosis y la hemostasis (23).
La deficiencia de PgI2 encontrada en la preeclampsia puede resultar en la sensibilización de la angiotensina II. La alteración del balance entre PgI2 y TxA2 contribuiría a una mayor reactividad plaquetaria y a daño vascular. En el feto se reduce la producción de PgI2 en los vasos del cordón umbilical. La placenta forma más TxA2 y menos PgI2. Las arterias umbilicales no responden al estímulo para producir PgI2 (24).
La PgI2 puede inducir la secreción de angiotensina II uteroplacentaria, que aumenta la presión y mejora la perfusión, lo cual es un estímulo extra para la secreción de PgI2 y NO por los vasos uteroplacentarios. Es decir, las necesidades del feto se cubren a costa del incremento de la presión arterial materna. Como tal, los efectos sobre la madre y el feto no se benefician con bajar la presión arterial con medicamentos, lo que debe ser tenido muy en cuenta cuando pensemos en utilizar agentes antihipertensivos en la preeclampsia.
La falla en producir PgI2 en respuesta al estímulo fisiológico, puede resultar en mayor resistencia de la arteria umbilical debida a vasoconstricción, especialmente por aumento en la producción de TxA2 por la placenta; tampoco existe reconocimiento de que hay daño vascular. Esta alteración del balance ha promovido el empleo de ácido acetilsalicílico para el tratamiento y la prevención de la preeclampsia, con beneficio limitado (25,26).
Los factores vasoconstrictores liberados por las células endoteliales son el péptido endotelina, el tromboxano A2 y los aniones superóxidos.
Endotelinas
Las endotelinas representan los factores vasoconstrictores más potentes existentes. Ejercen la acción vasoconstrictora vía receptores endotelina A (ETA) de los músculos lisos. Al actuar vía receptores ETB permiten que el endotelio libere NO y PgI2, inhibiendo la activación de las plaquetas.
Las endotelinas pueden liberar el activador de plasminógeno tisular (tPA) y aumentar la actividad fibrinolítica. Aumentan en la preeclampsia, en correlación con el factor von Willebrand y la fibronectina (27). Además, los niveles de endotelina se encuentran elevados en otras complicaciones obstétricas, como la insuficiencia orgánica múltiple de la preeclampsia grave, el síndrome HELLP (hemólisis, enzimas hepáticas elevadas, plaquetopenia), la hipertensión resistente al tratamiento, en el aumento del péptido natriurético atrial. Su elevación se asocia a una menor síntesis de PgI2 tisular en los casos de hipertensión, insuficiencia placentaria, RCIU, disfunción renal.
La alteración de la invasión trofoblástica y la consecuente alteración de la perfusión placentaria liberarían factores que activan la célula endotelial universalmente, originando la disfunción multisistémica de la preeclampsia. Este factor puede ser liberado por la placenta o por el neutrófilo activado y desaparece con el parto. La incubación de las células endoteliales de las mujeres con preeclampsia in vitro aumenta los niveles de fibronectina celular, que media la agregación y activación de las plaquetas (28). La lenta normalización de la fibronectina circulante refleja la también lenta recuperación del daño endotelial en la preeclampsia, lo que tendría un papel en mantener alta la presión arterial en el puerperio. Por otro lado, la endotelina-1 declina a la normalidad en el tercer día posparto, por lo que no sería el mayor agente vasoconstrictor en la fisiopatología de la preeclampsia (29).
ENDOTELIO Y COAGULACIÓN
El endotelio también controla la coagulación, por expresión en superficie de la trombo-modulina, que se une a la trombina, disminuyendo en simultáneo su afinidad por el fibrinógeno y aumentando su habilidad en activar la proteína C. En efecto, la proteína C es un anticoagulante que inactiva al factor Va, al factor VIII y al inhibidor del activador del plasminógeno. También segrega el cofactor de la proteína C –la proteína S- y los nucleótidos de la adenina (vasodilatadores específicos)
Pero, el endotelio puede ser trombogénico, al activar el factor von Willebrand, que estabiliza al factor VIII y actúa como cofactor en la adherencia de las plaquetas. Segrega el factor activador de plaquetas, que es un mediador importante en la producción plaqueta/fibrina. Estos factores normalmente promueven la coagulación local y la reparación en el sitio de daño (28) (Tabla 2).
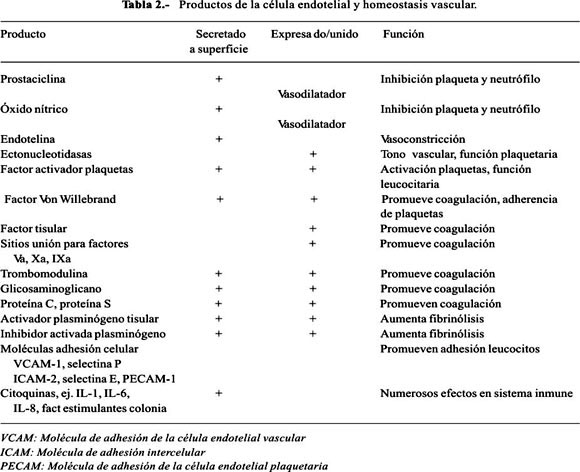
El endotelio puede actuar en la trombosis liberando el inhibidor del activador del plasminógeno 1 (PAI-1), que contrarresta los efectos fibrinolíticos del activador del plasminógeno de tipo tisular (t-PA) y previene o limita la coagulación y el daño vascular (18,30). Se ha observado que las células citotrofoblásticas aisladas de la placenta y del lecho placentario en los casos de restricción de crecimiento fetal (RCIU) expresan aumento de PAI-1, así como menor actividad del activador de plasminógeno, comparado con las células trofoblásticas en el embarazo normal. El aumento en la producción de PAI-1 restringiría la invasión endovascular del trofoblasto, muy temprano en el embarazo, al aumentar el depósito de fibrina y disminuir el flujo uteroplacentario en los embarazos con RCIU (31).
Normalmente, el endotelio, las plaquetas y los neutrófilos interactúan en homeostasis. La denudación del endotelio ocasiona trombosis. De manera similar, la disfunción del endotelio lo transforma de no trombogénico a una superficie trombogénica. También puede regular los neutrófilos y la adhesión y activación plaquetaria, ello por medio de la expresión y secreción de moléculas de adhesión celular.
La coagulación sanguínea consiste en una serie compleja de eventos que culminan en una fibrina insoluble. El sistema enzimático fibrinolítico es el mecanismo fisiológico que extrae esta fibrina. La alteración del balance puede acarrear trombosis. En la preeclampsia se activa la cascada de coagulación asociada a la activación del sistema fibrinolítico (32).
La adaptación anatómica, fisiológica y bioquímica en el embarazo normal ayuda a la madre al reto hemostático de la separación placentaria en el tercer período del parto; es decir, favorece la hipercoagulabilidad. La hipercoagulabilidad es también estimulada en la preeclampsia y eclampsia por disfunción de la célula endotelial, en respuesta a factor o factores desconocidos. Así, puede originar coagulación intravascular diseminada (CID), compensada o no, responsable de gran morbimortalidad materna y fetal (33).
Se ha encontrado que la activación de una nueva protrombinasa procoagulante, la protrombinasa fg12, por citoquinas tipo Th1, puede ocasionar aborto o preeclampsia y/o RCIU. El estrés o la acción de la endotoxina y de antígenos que ocasionan una respuesta de la citoquina Th1 aumentan los niveles de protrombinasa Fg12 en el trofoblasto y la decidua, resultando en aborto espontáneo de los embriones inmunogenéticamente "más débiles" (34,35).
ACTIVACIÓN DE LOS NEUTRÓFILOS
Los neutrófilos activados liberan sustancias que pueden mediar el daño vascular, incluyendo el contenido de los gránulos de neutrófilos, como las elastasas y otras proteasas. Se liberan especies de oxígeno tóxicas, que producen peroxidación lipídica de membrana, lisis de células endoteliales y aumento de la permeabilidad y reactividad vascular (2).
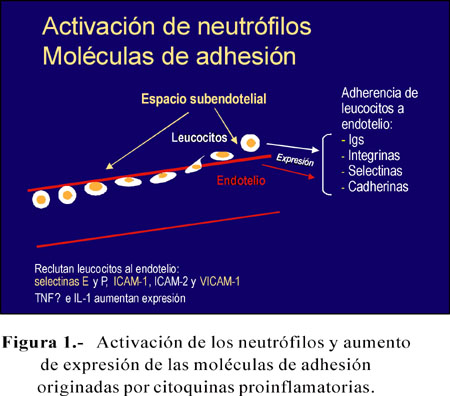
Cuando los neutrófilos son activados, ruedan por la superficie endotelial de la pared vascular y, en sitios específicos, se aplanan para pasar al espacio subendotelial. La adherencia de los neutrófilos al endotelio es mediada por moléculas de adhesión celular expresadas en el endotelio y en los leucocitos circulantes; en estas moléculas se encuentran las de la superfamilia de las inmunoglobulinas, las integrinas, selectinas, cadherinas (36).
En la superficie endotelial, las moléculas de adhesión celular que reclutan los leucocitos al endotelio son la selectina E y la selectina P, las moléculas de adhesión celular 1 y 2 (ICAM-1, ICAM-2) y la molécula de adhesión celular del endotelio vascular 1 (VICAM-1). La selectina E y las ICAM-1 y VCAM-1 se expresan nada o poco en las células endoteliales, pero aumentan por acción de las citoquinas proinflamatorias TNFa e IL-1 (37-40).
Las citoquinas proinflamatorias (IL-6, TNFa, la antagonista del receptor de IL-1 –que contrarresta a la IL-1) pueden activar los leucocitos y aumentar la expresión de las moléculas de adhesión celular en las células endoteliales (Figura 1).
Existe aumento de la producción de superóxidos de los neutrófilos, que pueden inhibir la síntesis de ciclooxigenasa y de sintasa PgI2. El aumento de las especies reactivas de O2 puede reorientar la vía del ácido araquidónico celular de la PgI2 a TxA2. Además, disminuyen los antioxidantes (41,42).
La alteración de la apoptosis de los neutrófilos puede explicar la neutrofilia asociada al embarazo normal. En las preeclámpticas, los neutrófilos activados permanecen en la circulación y pueden contribuir a la persistencia de la preeclampsia posparto. Los neutrófilos parecen modular la variación en la respuesta materna (43).
Como resumen sobre la activación de los neutrófilos, podemos decir que factores producidos por la placenta pueden activar los neutrófilos, al aumentar la generación de superóxidos y modular la expresión de las moléculas de adhesión. El incremento de expresión de la molécula de adhesión de superficie CD11 puede ser responsable de la mayor adhesión neutrófilo-endotelio inducida por factores derivados de las placentas en las mujeres con preeclampsia (44).
LÍPIDOS, DAÑO ENDOTELIAL Y PREECLAMPSIA
Además, las citoquinas que actúan sobre las células endoteliales o son liberadas por ellas pueden afectar muchos otros aspectos de la función del endotelio. Las células endoteliales pueden modificar el colesterol LDL, originando LDL oxidado dañino. Y las células endoteliales dañadas son fuentes de factores de crecimiento (18).
El metabolismo lípido anormal puede tener rol en la etiopatogenia. Hay aumento de los peróxidos lipídicos, lo que puede inhibir la síntesis PgI2, pero no de TxA2. También hay aumento de autoanticuerpos a LDL oxidado. Los vasos deciduales muestran necrosis fibrinoide de la pared vascular y acumulación focal de macrófagos cargados de lípidos -similar a lo que ocurre en la ateroesclerosis-, por peroxidación lipídica (45-47).
El plasma de las mujeres con preeclampsia puede alterar la relajación del miometrio vascular, actividad dependiente de endotelio. Una relación entre la apolipoproteína A1 y el comportamiento endotelial respalda la idea de que el metabolismo lípido alterado pueda estar involucrado en la disfunción endotelial característica de la preeclampsia (48),
Se ha visto que la melatonina protege la producción de NO en el endotelio de las arterias umbilicales contra la inhibición inducida por el LDL oxidado, seguramente por su habilidad de eliminar los radicales hidroxilo (49).
SISTEMA RENINA-ANGIOTENSINA EN LA UNIDAD FETOPLACENTARIA
Las células endoteliales de las venas en el tejido villoso de placenta y del ombligo pueden regular el sistema renina-angiotensina de la unidad fetoplacentaria. En respuesta a condiciones de hipoxia, la unidad fetoplacentaria induciría la actividad de la enzima convertidora de angiotensina (ACE) en la placenta, lo que permitiría regular la circulación fetal (50).
INVASIÓN TROFOBLÁSTICA Y MODIFICACIÓN DE VASOS UTERINOS
La invasión trofoblástica y la modificación de vasos uterinos son características importantes en la placentación de los mamíferos. Las arterias del endometrio son invadidas por el trofoblasto y así se establece el flujo sanguíneo placentario definitivo (51).
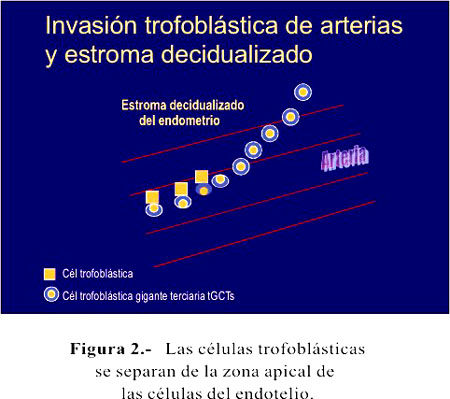
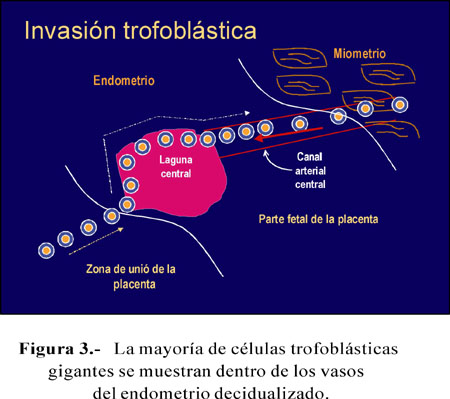
En la placenta en desarrollo del roedor, las células trofoblásticas gigantes terciarias (tGTCs) invaden profundamente las arterias y el estroma decidualizado del endometrio. Las células trofoblásticas, en su diferenciación a tGTCs, se separan de la zona apical de las células del endotelio (CE), se redondean y muestran un citoplasma muy basofílico y núcleos redondos (Figura 2). Estas tGTCs se muestran por primera vez a los 10 días posconcepción. Las tGTCs migran desde la zona de unión de la placenta a la superficie interna de la laguna central, migración que puede ser asimétrica, en un lado más rápido que en el otro (en el otro se ve endotelio libre de tGTCs). La migración se restringe inicialmente por la laguna central y más tarde por el canal arterial central. En los días subsiguientes, la migración de tGTC es a lo largo del canal arterial central de la parte fetal de la placenta a la zona de miometrio, es decir, va en sentido contrario al flujo sanguíneo arterial. La mayoría de tGTCs endovasculares se muestran en los vasos dentro del endometrio decidualizado y a tres cuartas partes de la interfase endometrio-miometrio. Pero, también pueden ser vistas en la parte miometrial de la arteria (Figura 3).
Desde el día 13 posconcepción, las tGTCs varían el endotelio de estos vasos. Mientras en la parte fetal de la placenta, las tGTCs pueden formar una multicapa en la arteria, en la parte materna de la placenta con más frecuencia forman una capa adosada a la pared interna de la arteria. En algunos casos, se ve grupos de tGTCs que llenan parcialmente el lumen arterial, pero no parecen afectar el flujo sanguíneo.
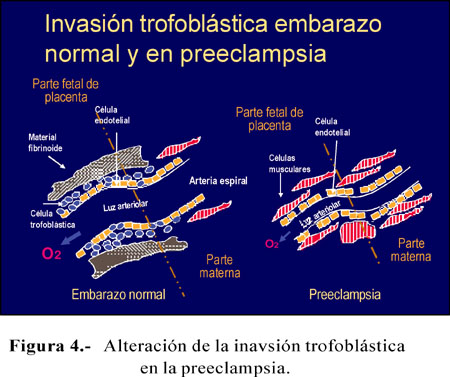
La placentación en la placenta hemocorial se caracteriza por invasión extensa de las células trofoblásticas en el endometrio. Las tGTCs invaden más profundamente. La invasión de la pared arterial trae cambios fisiológicos sustanciales para que haya un flujo sanguíneo apropiado a la placenta en desarrollo. Por otro lado, la ausencia de modificación de la pared arterial puede traer complicaciones del embarazo, como hipertensión, restricción del crecimiento del embrión, preeclampsia, entre otros (Figura 4).
Hay dos tipos de invasión de tGTC en la rata: a) la endovascular es la principal, que la realiza a lo largo de la superficie interna de la pared arterial, contra el flujo sanguíneo; y b) la intersticial, vía el espacio extracelular de la decidua basalis hacia el canal arterial central. En las placentas humana y del macaco existen ambos tipos, mientras en varias especies de roedores (ratón, rata, hamster) predomina la invasión tipo endovascular (51).
Invasión y respuesta inmune
No puede descartarse que la invasión profunda del endometrio por el trofoblasto prevenga la reacción de rechazo del embrión antigénico por el organismo materno. Probablemente, las células trofoblásticas invasoras producen hormonas que controlan la respuesta inmune del endometrio (51).
En la respuesta inmune activa en el embarazo hay mayor actividad de anticuerpos antiendoteliales (AECA). Ello sugiere el papel de los autoanticuerpos como factor que contribuye a la tolerancia fetal. La alteración de la regulación inmune, como la disminución de IgM-AECA en el suero de pacientes con lupus eritematoso sistémico (LES), puede contribuir a alterar la función reproductiva (52).
Adhesividad de célula trofoblástica
La capacidad de adhesividad es esencial para prevenir la descamación de la célula trofoblástica por el flujo sanguíneo contracorriente, así como, su diseminación por la sangre a sitios ectópicos del organismo materno. Existe cadherina-E en los trofoblastos que se diferencian a fenotipo invasivo. Se ve cadherina-E en los citotrofoblastos villosos y trofoblastos intermedios que no proliferan (IT), en las columnas e islas celulares de las placentas intrauterina, ectópica y en la mola parcial. Los trofoblastos que se transforman en mesénquima epitelial (EMT) y se separan de las columnas de células distales y de los trofoblastos extravillosos intersticiales (EVT) aislados más profundos no tienen cadherina-E. Los trofoblastos intraluminales, endovasculares y perivasculares adyacentes a los vasos maternos son también cadherina-E positivos, pero con un patrón muy variable según la edad gestacional. Esta variación temporal en la expresión de cadherina-E en los trofoblastos extravillosos les permite tener un potencial migrante e invasivo. La cadherina-E funcional puede restaurarse cuando los trofoblastos se agregan en la decidua y en la pared vascular, al completar su migración (53).
Factores angiogénicos
El desarrollo de la villosidad placentaria requiere la acción coordinada de factores angiogénicos en las células endoteliales y trofoblásticas. Un símil al factor de crecimiento del endotelio vascular (VEGF), el VEGF-C, aumenta la permeabilidad vascular y la proliferación/migración de la célula del endotelio. Existen receptores de VEGFR-2 y VEGFR-3 funcionales en el trofoblasto. La disminución en la expresión de VEGF-C y VEGFR-3 contribuirían al desarrollo anormal de la villosidad en la placenta de fetos RCIU (54-56).
El factor de crecimiento endotelial vascular (VEGF) induce aumento de la PgI2, pero no de NO, Las células estimuladas con plasma de gestantes con preeclampsia aumentan la producción de PgI2 y NO. El incremento en PgI2 por el suero de mujeres con preeclampsia no ocurre en presencia de anticuerpo anti-VEGF. El VEGF altera la función de la célula endotelial de manera análoga a la del plasma de mujeres con preeclampsia (57,58).
CONCLUSIONES
La preeclampsia no es sólo una hipertensión inducida por el embarazo, sino es secundaria a interacciones que provienen de una perfusión placentaria disminuida así como de la alteración en la función endotelial materna. La contribución materna es de factores que anteceden al embarazo, influenciados por las adaptaciones metabólicas usuales. El endotelio y otros blancos de los efectos de estas interacciones son más sensibles a las grandes modificaciones del embarazo, por activación de la cascada inflamatoria normal del embarazo.
Parte de la respuesta a la disminución de la perfusión placentaria puede ser por adaptación del feto a la menor cantidad de nutrientes recibidos, lo que debe tenerse en cuenta al elegir medicamentos a ser usados, así como, nos obliga a un seguimiento meticuloso del feto y del recién nacido. El punto de convergencia de esta interacción es a nivel del estrés oxidativo.
No hay un gen único que explique la preeclampsia, pero conocer la predisposición materna permite prevenir la preeclampsia en un grupo de mujeres (1,59).
Dada la prevalencia importante de la enfermedad, creo que es necesario que representantes de las diversas instituciones maternoperinatales del país nos sentemos a revisar la realidad de la preeclampsia-eclampsia en nuestro medio, incluyendo la verdadera incidencia de esta entidad, ya que un reciente estudio prospectivo nacional en Ecuador encontró una incidencia de preeclampsia y eclampsia de 22% en dicho país (60), lo cual duplicaría los hallazgos de algunos puntos del Perú (Tabla 1).
Espero contribuir con esta revisión a los avances en el conocimiento de la fisiopatología biomolecular de la preeclampsia.
BIBLIOGRAFÍA
1. Pacheco J. Enfermedad hipertensiva. En: Pacheco J (Ed). Ginecología y Obstetricia. 1ª edición. Lima, Perú. [ Links ]
2. Pipkin FB. Fortnightly Review: The hypertensive disorders of pregnancy. BMJ 1995; 311: 609-13. [ Links ]
3. Lansac J, Gardeil F. Hypertensive disorders in pregnancy. University Hospital, Tours-France, Coombe Womens Hospital, Dublin-Ireland. Internet 20 ago 2000. [ Links ]
4. Farro A. Comunicación personal, 31 marzo 2003.
5. Calderón N, Carvajal R, Herrera C, Ñique C. Frecuencia de la hipertensión en el embarazo. Ginecol Obstet (Perú) 1997; 43(1): 29-32. [ Links ]
6. Purizaca M. Evolución de la eclampsia en el Hospital III EsSalud Cayetano Heredia de Piura. Experiencia en 24 años. Ginecol Obstet (Perú) 1999; 45(4): 262-9. [ Links ]
7. Távara L. Estado actual de la mortalidad materna en los hospitales del Perú. Ginecol Obstet (Perú) 1999; 45(1): 38-42. [ Links ]
8. Román-Pilco C, Román-Loayza C. Estudio comparativo entre el síndrome HELLP y el HELLP parcial. Ginecol Obstet (Perú) 2000; 46(2): 141-7. [ Links ]
9. Pacora P. Comunicación personal, 27 marzo 2003.
10. Huertas E. Comunicación personal, 28 marzo 2003.
11. Pacheco J, Valdivia E, Huaman M, Carrasco N, Yui L. Eclampsia: Experiencia en 30 años en el Hospital Nacional Edgardo Rebagliati Martins - IPSS Ginecol Obstet (Perú) 1989; 35: 10. [ Links ]
12. Román-Pilco C, Román-Loayza C. Eclampsia, mortalidad materna y perinatal. Hospital Nacional Cayetano Heredia. Ginecol Obstet (Perú) 1999; 45(4): 270-3. [ Links ]
13. Tapia C, Márquez M, Casas A. Síndrome HELLP en la altura. Ginecol Obstet (Perú) 1997; 43(1): 65-8. [ Links ]
14. Arana C, Donayre A. Síndrome HELLP. Ginecol Obstet (Perú) 2000; 46(3): 222-7. [ Links ]
15. Verhaar MC, Rabelink TJ. The endothelium: a gynecological and obstetric point of view. Eur J Obstet Gynecol Reprod Biol 2001 Feb; 94(2): 180-5. [ Links ]
16. Toth P. Clinical data supporting the importance of vascular LH/hCG receptors of uterine blood vessels. Semin Reprod Med 2001; 19(1): 55-61. [ Links ]
17. Ramón de Berrazueta J. Rol del calcio en regular el tono vascular normal y en la hipertensión arterial. Rev Esp Cardiol 1999; 52 Suppl 3: 25-33. [ Links ]
18. Lyall F, Greer IA. The vascular endothelium in normal pregnancy and pre-eclampsia. Rev Reprod 1996; 1: 107-16. [ Links ]
19. Knowles RG, Moncada S. Nitric oxide synthases in mammals. Biochem J 1994; 298: 249-58. [ Links ]
20. Myatt L, Brewer A, Brockman DE. The action of nitric oxide in the perfused human fetal-placental circulation. Am J Obstet Gynecol 1991; 164: 687-92. [ Links ]
21. Roberts J, (Magee-Womens Research Institute) Ferrell R (University of Pittsburgh Graduate School of Public Health). UPCM 1999; 11, No. 50.
22. Guo G, Lade JA, Wilton AN, Moses EK, Grehan M, Fu Y, Qiu H, Cooper DW, Brennecke SP. Genetic susceptibility to pre-eclampsia and chromosome 7q36. Human Genetics 1999; 105 (6): 641-7. [ Links ]
23. Rowe J, Campbell S, Gallery ED. Plasma from preeclamptic women stimulates decidual endothelial cell growth and prostacyclin but not nitric oxide production: close correlation of prostacyclin and thromboxane production. J Soc Gynecol Investig 2001; 8(1): 32-8. [ Links ]
24. Walsh SW. Pre-eclampsia: an imbalance in placental prostacyclin and thromboxane production. Am J Obstet Gynecol 1985; 152: 335-40. [ Links ]
25. Knight M, Duley L, Henderson-Smart DJ, King JF. Antiplatelet agents for preventing and treating pre-eclampsia (Cochrane Review). En: The Cochrane Library, Issue 3, 2000. [ Links ]
26. Duley L, Henderson-Smart D, Knight M, King J. Antiplatelet drugs for prevention of pre-eclampsia and its consequences: systematic review. BMJ 2001; 322: 329-33. [ Links ]
27. Greer IA, Leask R, Hodson BA, Dawes J, Kilpatrick DC, Liston WA. Endothelin, elastase and endothelial dysfunction in pre-eclampsia. Lancet 1991; i: 158. [ Links ]
28. Taylor RN, Casal DC, Jones LA, Varma M, Martin JN Jr, Roberts JM. Selective effects of preeclampsia sera on human endothelial cell pro-coagulant protein expression. Am J Obstet Gynecol 1991; 165: 1705-10. [ Links ]
29. Makkonen N, Heinonen S, Hongisto T, Penttila I, Kirkinen P. Normalization of vasoactive changes in preeclampsia precedes clinical recovery. Hypertens Pregnancy 2002; 21(1): 51-64. [ Links ]
30. Bloom AL, Forbes CD, Thomas DT, Tuddenham EGD, Eds. Haemostasis and Thrombosis. Singapore: Longman Singapore Publ Ltd. 1994. [ Links ]
31. Sheppard BL, Bonnar J. Uteroplacental hemostasis in intrauterine fetal growth retardation. Semin Thromb Hemost 1999; 25(5): 443-6. [ Links ]
32. Davies JA, Prentice CRM. Coagulation changes in pregnancy-induced hypertension and growth retardation. En Greer IA, Turpie AGG, Forbes CD (Eds). Haemostasis and Thrombosis in Obstetrics and Gynecology. London: Chapman and Hall. 1992: 43-162. [ Links ]
33. Jambhulkar S, Shrikhande A, Shrivastava R, Deshmukh K. Coagulation profile in pregnancy induced hypertension. Indian J Hematol Blood Transf 2001; 19(1) [ Links ]:
34. Knackstedt M, Ding JW, Arck PC, Hertwig K, Coulam CB, August C, Lea R, Dudenhausen JW, Gorczynski RM, Levy GA, Clark DA. Activation of the novel prothrombinase, fg12, as a basis for the pregnancy complications spontaneous abortion and pre-eclampsia. Am J Reprod Immunol 2001; 46(3): 196-210. [ Links ]
35. Clark DA, Ding JW, Chaouat G, Coulam CB, August C, Levy GA. The emerging role of immunoregulation of fibrinogen-related procoagulant Fgl2 in the success or spontaneous abortion of early pregnancy in mice and humans. Am J Reprod Immunol 1999; 42(1): 37-43. [ Links ]
36. Harlan JM, Liu DY (Eds). Adhesion: Its Role in Inflammatory Disease. NY:WH Freeman & Co. 1992. [ Links ]
37. Haskard DO. Adhesive proteins. En: Bloom AL, Forbes CD, Thomas DT, Tuddenham EGD (Eds). Haemostasis and Thrombosis. Singapore: Longman Singapore Publ. 1994: 233-57. [ Links ]
38. Heimrath J, Krawczenko A, Dus D. Increased maternal plasma levels of soluble vascular cell adhesion molecule-1 (VCAM-1) in pregnancy induced hypertension (PIH). Ginekol Pol 2000; 71(4): 247-50. [ Links ]
39. Oyama R. The relationship between the level of expression of intercellular adhesion molecule-1 in placenta and onset of preeclampsia. J Obstet Gynaecol Res 2001; 27(3): 147-54. [ Links ]
40. Lorant DE, Li W, Tabatabaei N, Garver MK, Albertine KH. P-selectin expression by endothelial cells is decreased in neonatal rats and human premature infants. Blood 1999; 94(2): 600-9. [ Links ]
41. Chen G, Wilson R, Cumming G, Walker JJ, Smith WE, McKillop JH. Prostacyclin, thromboxane and antioxidant levels in pregnancy induced hypertension. Eur J Obstet Gynecol Repr Biol 1993; 50: 243-50. [ Links ]
42. Roes EM, Raijmakers MT, Zusterzeel PL, Knapen MC, Peters WH, Steegers EA. Deficient detoxifying capacity in the pathophysiology of preeclampsia. Med Hypotheses 2000; 55(5): 415-8. [ Links ]
43. Von Dadelszen P, Watson RW, Noorwali F, Marshall JC, Parodo J, Farine D, Lye SJ, Ritchie JW, Rotstein OD. Maternal neutrophil apoptosis in normal pregnancy, preeclampsia, and normotensive intrauterine growth restriction. Am J Obstet Gynecol 1999; 181(2): 408-14. [ Links ]
44. Wang Y, Gu Y, Philibert L, Lucas MJ. Neutrophil activation induced by placental factors in normal and pre-eclamptic pregnancies in vitro. Placenta 2001; 22(6): 560-5. [ Links ]
45. Maseki M, Nishigaki I, Hagihara M, Tomoda Y, Yagi K. Lipid peroxide levels and lipid content of serum lipoprotein fractions of pregnant subjects with and without pre-eclampsia. Clim Chim Acta 1981; 115: 155-61. [ Links ]
46. Branch DW, Mitchell MD, Miller E, Palinski W, Witzum JL. Pre-eclampsia and serum antibodies to oxidised low-density lipoproteins. Lancet 1994; 343: 645-6. [ Links ]
47. Gratacos E. Lipid-mediated endothelial dysfunction: a common factor to preeclampsia and chronic vascular disease. Eur J Obstet Gynecol Reprod Biol 2000; 92(1): 63-6. [ Links ]
48. Hayman RG, Sattar N, Warren AY, Greer I, Johnson IR, Baker PN. Relationship between myometrial resistance artery behavior and circulating lipid composition. Am J Obstet Gynecol 1999; 180(2 Pt 1): 381-6. [ Links ]
49. Wakatsuki A, Okatani Y, Ikenoue N, Shinohara K, Watanabe K, Fukaya T. Melatonin protects against oxidized low-density lipoprotein-induced inhibition of nitric oxide production in human umbilical artery. J Pineal Res 2001; 31(3): 281-8. [ Links ]
50. Ito M, Itakura A, Ohno Y, Nomura M, Senga T, Nagasaka T, Mizutani S. Possible activation of the renin-angiotensin system in the feto-placental unit in preeclampsia. J Clin Endocrinol Metab 2002; 87(4): 1871-8. [ Links ]
51. Zybina TG, Zybina EV. Genome multiplication In the tertiary giant trophoblast cells in the course of their endovascular and interstitial invasion into the rat placenta decidua basalis. Early Pregnancy: Biology and Medicine 2000; 4(2): 099-109. [ Links ]
52. Mendonca LL, Khamashta MA, Cuadrado MJ, Bertolaccini ML, Hughes GR. Natural immune response involving anti-endothelial cell antibodies in normal and lupus pregnancy. Arthritis Rheum 2000; 43(7): 1511-5. [ Links ]
53. Floridon C, Nielsen O, Holund B, Sunde L, Westergaard JG, Thomsen SG, Teisner B. Localization of E-cadherin in villous, extravillous and vascular trophoblasts during intrauterine, ectopic and molar pregnancy. Mol Hum Reprod 2000; 6(10): 943-50. [ Links ]
54. Dunk C, Ahmed A. Expression of VEGF-C and activation of its receptors VEGFR-2 and VEGFR-3 in trophoblast. Histol Histopathol 2001; 16(2): 359-75. [ Links ]
55. Ahmed A, Perkins J. Angiogenesis and intrauterine growth restriction. Baillieres Best Pract Res Clin Obstet Gynaecol 2000; 14(6): 981-98. [ Links ]
56. Dunk C, Shams M, Nijjar S, Rhaman M, Qiu Y, Bussolati B, Ahmed A. Angiopoietin-1 and angiopoietin-2 activate trophoblast Tie-2 to promote growth and migration during placental development. Am J Pathol 2000; 156(6): 2185-99. [ Links ]
57. Brockelsby JC, Anthony FW, Johnson IR, Baker PN. The effects of vascular endothelial growth factor on endothelial cells: a potential role in preeclampsia. Am J Obstet Gynecol 2000; 182(1 Pt 1): 176-83. [ Links ]
58. Brockelsby J, Hayman R, Ahmed A, Warren A, Johnson I, Baker P. VEGF via VEGF receptor-1 (Flt-1) mimics preeclamptic plasma in inhibiting uterine blood vessel relaxation in pregnancy: implications in the pathogenesis of preeclampsia. Lab Invest 1999; 79(9): 1101-11. [ Links ]
59. Roberts JM, Lain KY. Recent insights into the pathogenesis of pre-eclampsia. Placenta 2002; 23(5): 359-72. [ Links ]
60. Calle A, Hernández D, Campuzano C. Hipertensión inducida por el embarazo. Tema Oficial N° 2. XV Congreso FESGO Manta 2002. Rev Ecuat Ginecol Obstet 2002; 9(2): 207-20. [ Links ]
Dr. José Pacheco Romero
Calle Venecia 225
Lima 41, Perú
E-mail: jpachecoperu@viabcp.com
