










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. v.65 n.1 Lima mar. 2004
Acidosis láctica severa y leucemia aguda
David Loja1, Maricela Vilca2, Ricardo Postigo1, Aimee Torres3, José Alvizuri4
1Departamento de Medicina Interna, Hospital Arzobispo Loayza.Lima-Perú. 2Centro Médico-Quirúrgico.
Clínica ESSALUD. 3Servicio de Hematología Clínica, Hospital Arzobispo Loayza. Lima-Perú.
4Departamento de Cirugía General, Hospital Arzobispo Loayza. Lima-Perú.
Resumen
Reportamos el caso de una paciente de 27 años de edad con leucemia linfoblástica aguda, quien presentó acidosis láctica severa como complicación metabólica. Ella acudió con desnutrición severa, anemia marcada y síndrome consuntivo. No había compromiso del sistema reticuloendotelial y un mielograma inicial fue normal. Estos factores retardaron el diagnóstico y obligaron a ampliar el diagnóstico diferencial. La sospecha de neoplasia hematológica asociada a acidosis láctica sin causa aparente permitió reevaluar el caso con un nuevo mielograma y establecer el diagnóstico.
Palabras clave: Acidosis láctica; leucemia aguda de célula-B; hexocinasa: factor de necrosis tumoral.
Severe lactic acidosis and acute leukemia
Summary
We report the case of a 27 year-old patient with acute lymphoblastic leukemia who presented severe lactic acidosis as a metabolic complication. She presented with severe malnutrition, marked anemia and consuming syndrome. There was no reticuloendothelial system compromise and an initial myelogram was normal. These factors delayed the diagnosis and forced the extent of the differential diagnosis. The suspicion of lactic acidosis-associated hematologic neoplasia needed reassessing the case with new myelogram to establish the diagnosis.
Key words: Acidosis, lactic; leukemia, B-cell, acute; hexokinase; tumor necrosis factor.
La acidosis láctica (AL) es una rara complicación metabólica de las neoplasias hematológicas, que resulta de una excesiva producción de lactato (dependiente del tumor) y/o una disminución en la eliminación de lactato (metabolización hepática y aclaramiento renal). Las células neoplásicas pueden producir exceso de lactato debido a su mayor tasa de glicólisis, probablemente por sobreexpresión de la enzima glicolítica hexokinasa (1).
Clínicamente, los pacientes con esta emergencia presentan náuseas, vómitos, dolor abdominal, diarrea, alteración del sensorio, deshidratación, hipotensión arterial y descompensación hemodinámica. Los parámetros de laboratorio incluyen un nivel de lactato sérico mayor a 2 mmol/L, pH arterial menor de 7,25, anion gap mayor a 22 mEq/l y relación lactato/piruvato 10/1 (1-3).
Reportamos el caso de una paciente con leucemia aguda asociada a acidosis láctica severa. Se revisa la literatura.
CASO CLÍNICO
Paciente mujer de 27 años de edad, natural de Jaén – Cajamarca, que ingresa al Hospital Arzobispo Loayza el 13 de enero de 2003 con un tiempo de enfermedad de 6 meses, caracterizada por hiporexia, náuseas, debilidad y disnea. Además, refiere edema de miembros inferiores que se extendía a las manos y la cara. Tres meses antes del ingreso le diagnosticaron bartonellosis y anemia severa cuantificada en 4,8 g%, por lo que le transfundieron 4 paquetes globulares y le prescribieron rifampicina durante 15 días. En los últimos 5 días de tratamiento, acusó náuseas y vómitos, que persistieron hasta el ingreso. Aproximadamente 45 días antes del ingreso notó dolor precordial, intolerancia al calor y sed incrementada, que se mantuvieron durante la hospitalización.
Funciones biológicas: Apetito muy disminuido. Sed, marcado incremento. Orina poliúrica. Deposiciones, constipación. Pérdida ponderal de 15 kg en 6 meses.
Antecedentes: Poliomielitis al año de edad. Menarquia a los 13 años. G1 P1001. Cesareada por estrechez pélvica.
Examen físico: Presión arterial 120/80 mm Hg, frecuencia cardiaca 106x min, frecuencia respiratoria 24x min, temperatura 37,4°C, índice de masa corporal 13,5.
Lúcida, orientada, mal estado general, mal estado de nutrición e hidratación. Tejido celular subcutáneo muy disminuido. Palidez de piel y mucosas. Aparato respiratorio: vibraciones vocales conservadas; murmullo vesicular audible en ambos campos pulmonares, sin ruidos agregados. Cardiovascular: Ruidos cardiacos rítmicos de buena intensidad, ausencia de soplos. Abdomen: Blando, depresible, no puntos dolorosos; ruidos hidroaéreos presentes. No visceromegalia. Genito-urinario: Puntos renoureterales negativos. Neurológico sin alteraciones.
Exámenes auxiliares:
14-01 Hemoglobina 8,6 g%, hematocrito 25,1%, plaquetas 140,000xmL, leucocitos 2900xmL, abastonados 2, segmentados 42, eosinófilos 7, monocitos 4, linfocitos 45, velocidad de sedimentación globular 74 mm (1a hora), 112 mm (2a hora). Frotís de sangre periférica: Hipocromía+, anisocitosis++, plaquetas normales, leucocitos algo disminuidos, reticulocitos 4,7%, hematocrito 26%, proteínas totales 7,34 mg/dL, albúmina 3,93 mg/dL, globulina 3,41 mg/dL. T. Protrombina: 11,7" (INR: 1).
15-1 Examen de Orina Completa: Densidad 1020, pH: 5, proteínas 2 g%, células epiteliales algunas, leucocitos 20-30/c algunos degenerados y aglutinados, hematíes 6-8/c, cilindros granulosos.
16-01 Transaminasa glutámica pirúvica: 20 U/L (0-37), transaminasa glutámica oxalacética 35 U/L (0-34), bilirrubinas totales 0,67 mg/dL, bilirrubina directa 0,20 mg/dL, lactato deshidrogenasa 344 U/L (240-480). Coombs directo Negativo. T3L: 1,35 pg/mL (1,4-4,4). T4L: 1,01 ng/dL (0,8-2,0). TSH: 1,87 uUI/mL (0,27-4,20). Ferritina: 917,8 ng/mL (13-150). Radiografía de tórax: No se encuentra anormalidades en los campos pulmonares ni en la silueta cardiovascular.
17-01 Ecografía de hemiabdomen superior, hígado y bazo de caracteres normales. Ecografía de hemiabdomen inferior normal. Urocultivo: E. coli > 100,000 unidades formadoras de colonias.
21-01 Mielograma: Médula ósea con poca celularidad, se observa conservación de pleomorfismo celular. No se observa alteraciones morfológicas en sus series celulares. Pancitopenia de probable origen periférico. Anticuerpos antinucleares negativo. Anti-ADN nativo negativo.
22-01 Proteinuria 24 h 1,036 g. Depuración de creatinina 54 mg/mL/min/1,7mc. Coproparasitológico seriado nNegativo. Thevenon en heces seriado negativo.
24-01 Factor reumatoide < 0,14 (normal < 0,14). Proteína C reactiva 0,38 mg/dL (0-0,5).
27-01 Urocultivo negativo. Aglutinaciones tíficas y paratíficas negativas. Set de Brucella negativo.
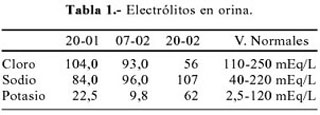
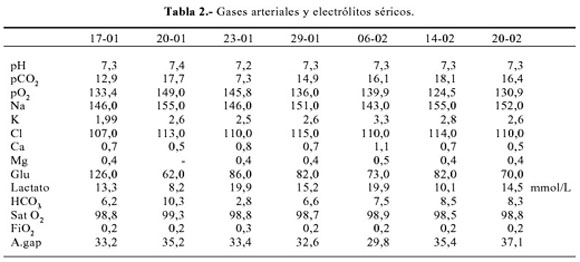
10-02 Proteinuria 24h 0,374 g. Velocidad de sedimentación globular 53 mm/1h. Aldosterona sérica (decúbito) 31,6 ng% (2,94 – 31,3) (Tabla 1 y 2).
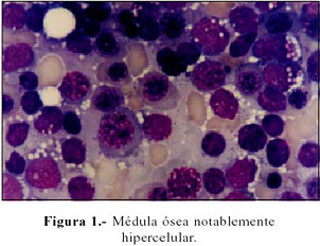
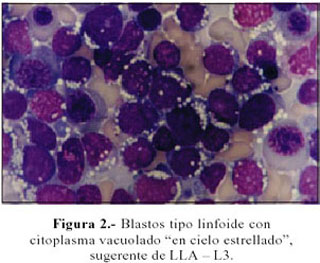
Evolución: Persiste con vómitos e hiporexia, intolerancia al calor y sed incrementada. Evoluciona subfebril y con pérdida ponderal. Se realiza un nuevo mielograma, el que se informa con 80% de blastos en relación a la celularidad medular total, compatible con leucemia aguda. Figura 1 y 2. Se decide su transferencia a Instituto Especializado, donde el aspirado de médula ósea ratifica la condición de leucemia linfática de células precursoras de estirpe B, con expresión de CD15, CD13 y CD33, a descartar bifenotipo. Recibe un curso de vincristina y prednisona y no acude a sus controles.
DISCUSIÓN
La acidosis láctica es un trastorno ácido-básico consecutivo a la acumulación del ácido láctico, el cual se comporta como la forma reducida del ácido pirúvico. El metabolismo de la glucosa a piruvato por la vía glucolítica se lleva a cabo en el citosol y no precisa oxígeno. El piruvato puede desdoblarse hasta dióxido de carbono y agua, por la vía del ciclo del ácido cítrico de Krebs, proceso aeróbico que ocurre en las mitocondrias. De este modo, la concentración del ácido pirúvico en el citosol refleja el equilibrio entre la producción por glucólisis y la degradación por oxidación mitocondrial (3,4).
Debido a que el piruvato es el único precursor del lactato, los factores que regulan su metabolismo también controlan los del lactato. Entonces los factores que regulan la concentración sanguínea del lactato son: el nivel sérico de piruvato, la concentración sanguínea de hidrogeniones y la relación nicotin adenín dinucleótido reducido/nicotín adenín dinucléotido (NADH/NAD+), que refleja el estado de oxidorreducción del citosol (3,4).
La producción normal de lactato es de unos 20 mmol/kg/día; la mitad deriva de la glucólisis. Aproximadamente,se producen a diario 1400 mmol de ácido láctico, los cuales son tamponados por 1400 mmol de bicarbonato a lactato de sodio. El hígado es el responsable para oxidar el lactato y restaurar el bicarbonato. Los riñones también contribuyen en la remoción de al menos de 10 a 20% del total de lactato metabolizado y lo hacen a través de tres vías: excreción, gluconeogénesis y oxidación. La excreción de ácido neto del organismo es de unos 70 mmol/día. Por consiguiente, la utilización del lactato tiene un papel crítico en su homeostasia normal (5).
Todos los órganos pueden producir y utilizar lactato en condiciones especiales, excepto los eritrocitos, que no lo usan por falta de mitocondrias. Los tejidos que producen exceso de lactato son: piel (25% de producción), eritrocitos (20%), cerebro (20%), músculo (25%), intestino (10%). Durante el ejercicio extenuante, el músculo esquelético contribuye con el mayor aporte de lactato circulante. Durante el embarazo, la placenta es un importante productor de lactato, el cual atraviesa hacia la circulación fetal (5,6).
Se considera hiperlactacidemia cuando se alcanza un nivel entre 2 a 5 mmol/L, y acidosis láctica severa si es mayor a 5 mmol/L, como el caso que discutimos. Niveles elevados y de larga duración, por encima de 5 mmol/L se asocian con tasas de mortalidad muy altas. Acidosis lácticas severas pueden cursar con leve incremento del anion gap, debido a que niveles de lactato (6-10 mmol/L) asociados con alta mortalidad, alteran poco el anion gap. En tanto que niveles muy altos (30 mmol/kg) de breve duración, como los ocasionados por ejercicio severo o convulsiones, no se asocian con mayor mortalidad (1,3).
Si bien es cierto que el ácido láctico se identificó por primera vez en los tejidos humanos a principios del siglo XIX y se le relacionó como mediador de la acidosis clínica a mediados de 1920, recién desde los 60, los clínicos apreciaron que el incremento de la concentración sérica del lactato significaba clínicamente problemas para los pacientes (7).
En 1976, Cohen y Woods desarrollaron un sistema de clasificación (Tabla 3), que divide la acidosis láctica en dos categorías: tipo A (hipoxia hística) y tipo B (sin hipoxia tisular), que a su vez comprende tres categorías relacionadas con trastornos sistémicos graves (B1), con fármacos o toxinas (B2) y con errores congénitos del metabolismo (B3) (7).

En el subtipo B1, se describe la AL como una muy rara complicación de las neoplasias hematológicas, es el caso de nuestra paciente. Tal vez el subregistro o el subdiagnóstico han incidido en el recuento de únicamente 29 casos asociados con linfoma y 25 casos asociados con leucemia en la literatura inglesa (1,5).
Como ya se ha señalado, el lactato producto final de la glicólisis anaeróbica, es metabolizada a glucosa por el hígado y por los riñones. Las células neoplásicas pueden mantener una alta tasa de glicólisis aún en presencia de oxígeno y de este modo aportan grandes cantidades de lactato. Este fenómeno es magnificado si el tumor alcanza la circulación sanguínea o si la cantidad de tiamina disponible es insuficiente. Existen algunos factores que permiten esta alta tasa de glicólisis, como una expresión aberrante o sobreexpresión de enzimas glucolíticas como la hexokinasa, la acción sistémica y paracrina del factor de necrosis tumoral (TNF), la proliferación celular y el compromiso de la función hepática (1,5,6).
En condiciones normales, la insulina regula la expresión de la hexokinasa. Sin embargo, los factores de crecimiento similares a insulina (IGFs) y sus receptores, los cuales son sobreexpresados por algunas células cancerosas, pueden simular muchas acciones de la insulina y además juegan un rol importante en controlar la proliferación y metabolismo de las células cancerosas. El IGF-I induce la expresión de la hexokinasa (1).
Por otro lado, la proliferación celular puede causar elevadas concentraciones de TNF y de lactato o de ambas y se desconoce si uno es secundario al otro. Del mismo modo, la alta frecuencia de compromiso hepático en linfoma y leucemia sugiere la posibilidad de una menor utilización de lactato por este órgano y su consiguiente acumulación (1).
Los pacientes con AL típicamente se presentan con hiperventilación y en ocasiones con hipotensión asociadas a taquicardia, debilidad muscular, vómitos, diarrea y estupor (8,9). En nuestra paciente se evidenció la mayoría de estas características. Los resultados que confirmaron el diagnóstico incluyeron lactato sanguíneo elevado, acidosis metabólica con anion gap elevado, los que se mantuvieron inalterables aun después de haberse controlado la infección urinaria y mejorado las pruebas de función renal.
Asimismo, se excluyó las causas más frecuentes de AL tipo A relacionadas a neoplasias, tales como insuficiencia circulatoria, sepsis y shock mediante la historia clínica, evolución y datos de laboratorio. También se hizo lo propio con causas comunes de AL tipo B, como infecciones, diabetes mellitus, falla renal, insuficiencia hepática, déficit de tiamina, fármacos y toxinas.
La tasa de letalidad es muy alta en estas neoplasias hematológicas (26/28 y 24/25 casos de linfoma y leucemia, respectivamente, fallecieron) y precisamente la aparición de acidosis láctica es un marcador de pobre pronóstico. La AL revierte cuando la quimioterapia produce citorreducción de la neoplasia, pero la resolución total de la AL depende de la respuesta del tumor subyacente, la que justamente tiene una alta tasa de recurrencia (1,10,11).
En cuanto al tratamiento de la AL, se puede usar buffers exógenos con la intención de normalizar el pH arterial y atenuar el efecto deletéreo de la acidosis. Sin embargo,la alcalinización ha mostrado potenciación en la producción de lactato en pacientes con acidosis láctica crónica inducida por neoplasias. Los fármacos usados para tratar la AL pueden agravar la condición. El bicarbonato de sodio incrementa la producción de lactato. El carbicarb es una mixtura de carbonato y bicarbonato de sodio, respectivamente, que tampona como el bicarbonato de sodio pero sin generación de dióxido de carbono. Los resultados de estudios en animales son prometedores, aunque los ensayos clínicos son escasos todavía. El dicloroacetato estimula la piruvato deshidrogenasa y mejora los valores de laboratorio, pero no así la tasa de sobrevida (5,9).
El único tratamiento efectivo para la AL tipo A es la cesación en la producción de ácido láctico mediante la mejoría en la oxigenación tisular. Estas medidas incluyen tratamiento del shock, restauración del volumen circulante, mejoría de la función cardiaca, resección de áreas isquémicas y mejoría de la sepsis (5,8,9).
El manejo de la AL tipo B no tiene la urgencia de tratar como el tipo A, debido a que la condición no se asocia con el problema de generar ATP. En el caso de la intoxicación por etanol, su metabolismo genera NADH y lleva a la conversión de piruvato a lactato más que a glucosa, generando una acidosis leve, a diferencia de lo que ocurre en la deficiencia de tiamina, en la que la glucosa no puede ser oxidada anaeróbicamente; acá, el peligro es la injuria del sistema nervioso central que se produce cuando se incrementa la glicólisis para suplir el Adenosín trifosfato, que no es reemplazado por la oxidación de cetoácidos. La AL inducida por drogas puede ser causada por fosforilación oxidativa o por interferencia con la gluconeogénesis, de modo que la acidosis es severa, pero con buen pronóstico si se neutraliza el exceso de hidrogeniones con bicarbonato de sodio, enlenteciendo la producción de hidrogeniones con insulina para el caso de AL inducida por metformina, acelerando el metabolismo de lactato con cloroacetato e incrementando la eliminación de la droga implicada por excreción renal (5,8,9).
En nuestro caso, como en los casos reportados, la alcalinización fue ineficaz. No obstante, este tratamiento puede ser necesario para revertir los efectos adversos cardiovasculares de la hiperlactacidemia, como la angina de pecho, las arritmias y la descompensación hemodinámica, eventos que ocurrieron en nuestra paciente.
Desafortunadamente, entre 10 y 15% del bicarbonato administrado es inmediatamente convertido a dióxido de carbono y si el paciente es incapaz de eliminarlo por hiperventilación, el pH no mejora significativamente, para cuyo efecto se hace necesario el uso de hemofiltración o hemodiálisis con baño de bicarbonato para normalizar el pH; pero el nivel de lactacidemia permanece inalterable hasta que la quimioterapia funcione (11-14).
Otra modalidad de terapia en AL es la administración de insulina con glucosa, lo cual incrementa la conversión de piruvato a acetil coenzima A y por consiguiente facilita la oxidación de lactato a piruvato, pero la mejoría suele ser discreta (15,16).
Otros problemas observados en el presente caso, además de la acidosis metabólica con anion gap elevado por lactacidosis, fueron la acidosis metabólica anion gap normal por probable acidosis tubular renal tipo 1, sugerido por la presencia de hipokalemia dependiente de pérdidas renales, la presencia de acidosis metabólica anion gap normal ocultada por la administración de bicarbonato con delta anion gap / delta bicarbonato que fluctuó entre 0,8 y 1, y la presencia de carga neta urinaria positiva en 2,5, que refleja una acidificación distal insuficiente para el grado de acidosis sistémica. Todo ello quedó pendiente de corroborarse con la prueba de cloruro de amonio.
La presencia de aldosterona alta puede darse como respuesta a contracción de volumen, sin embargo, aunque infrecuente existen síndromes como el de Bartter, que cursa con hiperaldosteronismo sin hipertensión arterial, o el de Gitelmann, que además de hipokalemia muestra hipomagnesemia.
En suma, un caso con múltiples problemas que ofreció dificultad diagnóstica por la solapada presentación de acidosis láctica de aparente causa incierta.
BIBLIOGRAFÍA
1. Sillos EM, Shenep JL, Burghen GA, Pui CH, Behm FG, Sandlund JT. Lactic acidosis: A metabolic complication of hematologic malignancies. case report and review of the literature. Cancer 2001; 92(9): 2237-46. [ Links ]
2. Kar M. Oncological emergencies. J Indian Ac Clin Med 2000; 5(1): 32-7. [ Links ]
3. Heredero M, Mena V, Riveron R. Acidosis láctica: Algunas consideraciones. Rev Cubana Pdiatr 2000; 72(3): 183-93. [ Links ]
4. Mizock BA, Falk JL. Lactic acidosis in critical illness. Crit Care Med 1992; 20: 80-93. [ Links ]
5. Luft F. Lactic acidosis update for critical care clinicians. J Am Soc Nephrol 2001; 12: 815-9. [ Links ]
6. Keung Chau W, Fen Yang CH, Hong Chou Y, Hung Ho CH. Aggressive undifferentiated carcinoma of unknown primary site complicated by lactic acidosis after bleeding: A case report. Jpn J Clin Oncol 2002; 32(6): 210-4. [ Links ]
7. Cohen RD, Woods HF. Clinical and biochemical aspects of lactic acidosis. Roston Oxford Blackwell scientific publication; 1976: 1-200. [ Links ]
8. Frommer JP. Acidosis lactica. Intensive Care Med 1998; 81(4): 304-5. [ Links ]
9. Borron SW, Megarbane B. Lactic acidosis. www.emedicine.com al 10-10-03. [ Links ]
10. Doolitle GC, Wurster MW, Rosenfeld CS, Bodensteiner DC. Malignancy-induced lactic acidosis. South Med J 1988; 81(4): 533-6. [ Links ]
11. Ali AA, Flombaum CD, Brochstein JA, Gillio AP, Bussel JB, Boulad F. Lactic acidosis and renal enlargement at diagnosis and relapse of acute lymphoblastic leukemia. J Pediatr 1994; 125(4): 584-6. [ Links ]
12. Grossman L, Holloway R, DAcosta M, Roncari D, Lazarovits A, Baker M, et al. Lactic acidosis in a patient with acute leukemia. Clin Invest Med 1983; 6(2): 85-8. [ Links ]
13. Revesz T, Obeid K, Mpofu C. Severe lactic acidosis and renal involvement in a patient whit relapsed burkitts lymphoma. Pediatr Hematol Oncol 1995; 12(3): 283-8. [ Links ]
14. Aramideh M, Koopmans Rp, Koelman Jh, Stam J. Rapidly progressing peripheral neuropathy with lactic acidosis and coproporphyria in a patient with HTLV-1 associated t-cell leukaemia treated with zidovudine. J Neurol 2001; 248(7): 621-2. [ Links ]
15. Ishibashi M, Kimura N, Kawara T, Morioka E, Hisano S, Okomura M. Lactic acidosis complicating adult t-cell leukemia: Report of two cases. Eur J Hematol 1993; 50(2): 122-3. [ Links ]
16. Schlaifer D, Rigal-huguet F, Laurent G. Chronic lactic acidosis and acute lymphoblastic leukemia. Rev Med Interne 1990; 11(1): 91. [ Links ]
Dr. David Loja Oropeza
Reynolds 114-101
Lima 41, Perú.
E-mail: davidloja@hotmail.com

