










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. v.65 n.4 Lima oct./dic. 2004
Histiocitosis maligna
Reporte de un caso
Oscar Ruiz1,2, Willy Quiñones2, Oscar Misad3, Carlos Delgado1,2,
Sergio Ronceros1,4, Manuela Marangoni5, Luz Bardales2, Rafael Reyes2,
Alfredo Castillo2, Katia Urrutia2,
1Instituto de Investigaciones Clínicas, UNMSM. 2Servicio Hematología Clínica, Hospital Dos de Mayo.
3Laboratorio de Anatomia Patológica Oscar Misad. 4Departamento de Patología Clínica y
Anatomía Patológica, Hospital Dos de Mayo. 5Departamento de Enfermería, Hospital Dos de Mayo.
Resumen
Paciente varón de 14 años, procedente de Huancayo, que presenta un mes antes de su hospitalización intolerancia gástrica, ictericia, fiebre y síndrome linfoproliferativo. Se encontró en los exámenes de laboratorio pancitopenia severa por hemofagocitosis. El diagnóstico hematológico y anatomopatológico fue histiocitosis maligna. Se presenta este caso por ser una entidad infrecuente y una emergencia médica.
Palabras clave: Histiocitosis maligna; pancitopenia; hepatomegalia; esplenomegalia.
Malignant histiocytosis. Case report
Abstract
Fourteen year-old male patient referred from Huancayo who presented one month gastric intolerance, jaundice, fever and a lymph proliferative syndrome. Laboratory tests revealed severe pancytopenia due to phagocytosis. Haematologic and anatomy-pathology diagnosis was human malignant histiocytosis. We present this case due to its low frequency and the emergency character of the disease.
Key words: Histiocytosis, malignant; pancytopenia; hepatomegaly; splenomegaly.
INTRODUCCIÓN
El término de histiocitosis maligna fue usado por Rappaport para definir una enfermedad sistémica maligna caracterizada por la proliferación de histiocitos malignos y sus precursores. En la actualidad, el término involucra las proliferaciones malignas de las células terminales de la diferenciación monocítica, es decir, histiocitos y células dendríticas (1-11).
La histiocitosis maligna es un cuadro infrecuente, que puede presentarse a cualquier edad y sin predominio sexual. Clínicamente se manifiesta con fiebre, adenopatías, hepato-esplenomegalia y mal estado general (1-4,12-16).
Los exámenes de laboratorio detectan pancitopenia, hiperbilirrubinemia, alteraciones de las pruebas hepáticas y de la deshidrogenasa láctica. El curso de la enfermedad es rápidamente progresivo y a veces fulminante, con presencia de derrame pleural, ictericia, púrpura, pancitopenia grave y una supervivencia inferior a los 9 meses (1-3,5,9,12-15).
En sangre periférica, además de las citopenias, se puede observar histiocitos atípicos. En médula ósea se halla una infiltración por histiocitos de intensidad variable, en diferentes estadios de diferenciación; siendo el patrón infiltrativo medular de tipo intersticial, lo que permite que la médula ósea conserve su arquitectura (1,5-7,11,13).
La histiocitosis maligna también afecta ganglios, bazo e hígado, observándose inclusive actividad fagocítica (1-4,9,12,13,16).
Bryne y Rappaport resaltaron la importancia de identificar histiocitos atípicos con hemofagocitosis. En estudios posteriores, este hallazgo no es considerado necesario para el diagnóstico de la entidad. A diferencia del síndrome hemofagocítico reactivo, la fagocitosis en la histiocitosis maligna es un hecho poco llamativo y se la observa habitualmente en los histiocitos maduros (1,5,6,10,11).
Al inicio, los criterios diagnósticos de la histiocitosis maligna fueron morfológicos; después se ha ampliado a las técnicas de citoquímica, inmunohistoquímica, microscopía electrónica, citometría de flujo y biología molecular. El diagnóstico de histiocitosis maligna se debe basar en la identificación de histiocitos y la demostración de marcadores linfoides B y T (1,2,4-7,11).
El diagnóstico diferencial se debe hacer con linfomas anaplásicos de células grandes Ki-1 positivos y T periféricos, enfermedad de Hodgkin, histiocitosis reactivas -sobre todo a infecciones vírales-, siendo la diferenciación más difícil con la primera entidad. Los histiocitos neoplásicos no expresan grupo de diferenciación (CD) 30, a diferencia de los tumores Ki-1 positivos (1,6,15).
La evolución de la enfermedad es rápidamente progresiva y, sin tratamiento, el enfermo fallece en 2 a 6 meses. El pronóstico depende del grado de infiltración y disfunción orgánica, sobre todo en la médula ósea, hígado y pulmones.
El tratamiento es difícil de generalizar, ya que es una neoplasia rara. En los casos presentados se ha usado quimioterapia en los linfomas de grado alto de malignidad (ciclofosfamida, doxorrubicina, vincristina y pregnisona: CHOP-bleomicina). Algunos autores han utilizado, además, etopósido y arabinósido de citocina. Algunos autores recomiendan efectuar profilaxis del sistema nervioso central. El tratamiento de mantenimiento no ha demostrado utilidad (1,2,15).
HISTORIA CLÍNICA
Paciente varón de 14 años de edad, transferido del Hospital Regional de Huancayo, por presentar desde un mes antes distensión abdominal, alza térmica, astenia, náuseas y vómitos. Ocho días antes del ingreso, se agrega edema de miembros inferiores. Cinco días antes aparece ictericia en escleras y piel, presencia de tumoraciones en hipocondrio derecho y fosa ilíaca izquierda. Recibió transfusión de un paquete globular, furosemida y ampicilina.
Al examen clínico, el paciente se presenta: febril, hipotenso, polipneico, taquicárdico, adelgazado, con ictericia en piel y escleras, gingivorragia, púrpura generalizada y edema en miembros inferiores. Además, ganglio cervical derecho de 0,5 cm de diámetro (consistencia dura y adherido a planos profundos), abdomen distendido con edema de pared y hepato-esplenomegalia.
EXÁMENES AUXILIARES:
02-07-2003: Hemograma: hemoglobina 5,6 g/dL, hematocrito 17,5%, volumen corpuscular medio 97,8 fl, hemoglobina corpuscular media (HCM) 31,3 pg, concentración de HCM 32%, plaquetas: 20 000 x mL, leucocitos: 890 x mL, eosinófilos 1%, segmentados 38%, linfocitos 58%, monocitos 3%. Observaciones: anisocitosis ++, poiquilocitosis ++, granulaciones tóxicas +, policromatofilia +, dianocitos +, linfocitos atípicos 30%, linfocitos en flor 10%, macroplaquetas +; velocidad de sedimentación globular 40 mm/h, reticulocitos 0,6%.
Perfil de coagulación: tiempo parcial de tromboplastina 58,4"/33", tiempo de protombina 28,5"/13,5", índice internacional de normalización (INR) 2,54, tiempo de sangria 415", bilirrubina total 5,12 g/dL, bilirrubina directa: 5,00 g/dL, bilirrubina indirecta 0,12 g/dL, transaminasa glutámico-oxalacética (TGO) 341 U/L, transaminasa glutámico-pirúvica (TGP) 150 U/L, deshidrogenasa láctica (LDH) 1930 U/L, creatinina 0,5 mg/dL. Examen de orina: sangre + hematíes 18 a 20 x campo. Electrólitos: Na 128 mol/L, K 4,40 mol/L, Cl 103 mol/L, Ca 4,36 mol/L. Fosfatasa alcalina 1978,0 U/L. Aglutinaciones tífica, paratífica y brucela negativos. Cultivos negativos. Bacilo Koch (BK) en esputo inducido negativo. Gota gruesa (-), pruebas virales (toxoplasma, rubeola, citomegalovirus, virus del herpes (TORCH), virus del SIDA, hepatitis B, hepatitis C, virus linfoadenotrópico) negativas.
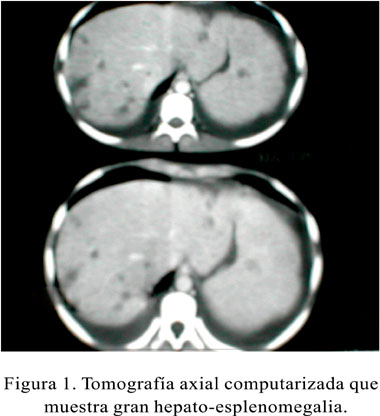
Ecografía: hepato-esplenomegalia moderada a severa, ascitis, derrame pleural derecho. Tomografía abdominal: hepatoesplenomegalia, abscesos hepáticos-esplénico, derrame pleural bilateral, ascitis laminar (Figura 1).
Mielograma: médula ósea con panfagocitosis e incremento de la serie linfoide, compatible con síndrome hemofagocítico, a descartar histiocitosis maligna. Citometría de flujo concluye: poblaciones linfoide T, B y mieloides de características normales; no se detecta proliferaciones clonales. Marcador CD 30 negativo.
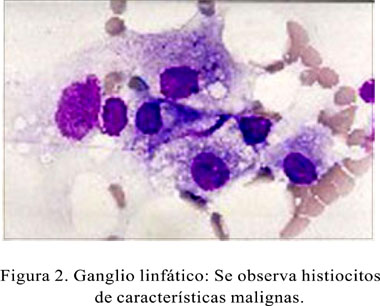
Biopsia de ganglio linfático: marcada dilatación de los sinusoides, los cuales están ocupados por histiocitos malignos, algunos de los cuales fagocitan hematíes y hemosiderina; hay además cambios en relación con mielopoyesis focal (Figura 2).
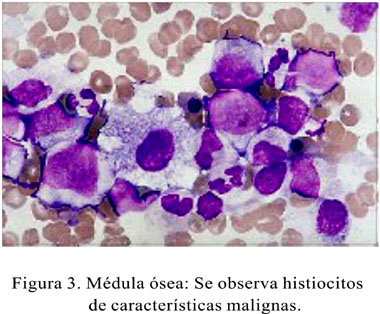
Biopsia de medula ósea: Extensamente necrosada, infiltrada difusamente por histiocitos malignos con las mismas características descritas en el ganglio. Hay además elementos residuales de la médula ósea constituidos en su mayor parte por elementos precursores con predominio de la serie roja (Figura 3). Conclusión: histiocitosis maligna que compromete ganglio y médula ósea.
Durante su hospitalización, el estado general del paciente se fue comprometiendo progresivamente hasta presentar insuficiencia respiratoria severa, que ocasionó la muerte.
DISCUSIÓN
En los últimos años, se ha observado una creciente complejidad en el diagnóstico histopatológico de las neoplasias linfohemáticas, especialmente relacionada con el impacto del desarrollo tecnológico (1,2,6-8) aplicado a la observación de las entidades analizadas comúnmente por cualquier patólogo práctico. Así, desde la época de Rappaport a la actualidad, frecuentemente se ha reformulado y reinterpretado diferentes cuadros, con el afán de lograr aislar entidades anatomoclínicas que sinteticen un conocimiento más acabado de su naturaleza, histogénesis, presentaciones epidemiológicas y agentes etiológicos asociados (1,16); conjunto de factores, que permitirían una proyección hacia una mejor comprensión del pronóstico y manejo terapéutico de estos tumores.
El caso que se reporta es infrecuente en la práctica médica. Se presenta como problema diagnóstico ante la similitud del cuadro hemofagocítico en la patología infecciosa propia de nuestro medio (1,9,12-14,16).
Se analizó los exámenes auxiliares y se tuvo en cuenta los datos que aportaban: pancitopenia grave en hemograma, con presencia de linfocitos atípicos y granulaciones tóxicas; LDH muy alta; ecografía abdominal con hepatoesplenomegalia, ascitis, sin presencia de ganglios intraabdominales y derrame pleural; tomografía axial computarizada abdominal con hepatoesplenomegalia, tumoraciones hepatoesplénicas, derrame pleural y ascitis; mielograma mostró la presencia de fagocitosis medular, granulaciones tóxicas, incremento de la serie linfoide y un conteo de blastos dentro de los parámetros normales. Estos hallazgos descartan la posibilidad de leucemia aguda, quedando como presunciones diagnósticas linfomas agresivos con invasión a medula ósea, síndromes hemofagocíticos, tanto de causa neoplásica como infecciosa (1,4,9,12-15).
La etiología infecciosa es descartada por la negatividad de todos los cultivos y los exámenes disponibles para encontrar una etiología viral. Sabemos que en las neutropenias febriles no siempre se encuentra la causa del cuadro. Por lo tanto, el espectro de las entidades nosológicas se va reduciendo a cuadros neoplásicos, como: linfomas malignos agresivos e histiocitosis maligna por la marcada hepatoesplenomegalia, ictericia, pancitopenia, fiebre, síndrome consuntivo y la presencia de linfocitos, monocitos atípicos en sangre periférica y fagocitosis medular. Por ello se plantea realizar procedimientos más invasivos, como biopsia de ganglio (a pesar del tamaño y ser único) y biopsia de médula ósea (1,4,6,8,10).
El diagnóstico de linfoma fue descartado por la ausencia de compromiso medular y por la citometría de flujo, cuyo resultado indicó que la población linfoide B y T eran normales en cantidad y en características, ausencia de clonas y marcador CD 30 negativo (2,3,6,8). Por todo lo anterior, el diagnóstico definitivo corresponde a un caso de histiocitosis maligana.
Se ha escogido este caso debido a que los cuadros de histiocitosis maligna son infrecuentes; pero, deben de ser tomados en cuenta en los pacientes con gran compromiso del estado general, fiebre, síndrome linfoproliferativo (hepatoesplenomegalia y adenomegalia), ictericia, daño de la función hepática y pancitopenia. El estudio de este caso ha permitido revisar la serie de entidades que producen síndromes linfoproliferativos asociados a hemofagocitosis; asimismo, plantear la necesidad de realizar procedimientos invasivos en pacientes con mal estado general, dándoles un margen de seguridad con la transfusión de los hemoderivados correspondientes. El estudio multidisciplinario puede aportar mayor cantidad de experiencia y conocimiento para el diagnóstico y manejo del paciente.
REFERENCIAS BIBLIOGRÁFICAS
1. Sans J. Hematología Clínica. 4ª ed. Madrid: Elsevier Science; 2001. p. 547-64. [ Links ]
2. Jaffe ES, Harris NL, Stein H, Vardiman J, eds. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues: WHO Classification of Tumours. Lyons: IARC Press; 2001. p. 230-5, 273-89. [ Links ]
3. Johnston RB Jr. Monocytes and macrophages. N Engl J Med. 1998;318:747-52. [ Links ]
4. Komp DM, Perry MC. The histiocytic syndromes. Semin Oncol. 1991;18:1-62. [ Links ]
5. Lipton JM. Histiocytic disorders. En: Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohe HJ, Silberstein LE, et al, (eds.). Hematology. Basic principles and practice. 3a ed. Nueva York: Churchill Livingstone; 2000. p. 783-96. [ Links ]
6. Favara BE, Feller AC, Pauli M, Jaffe ES, Weiss LM, Arico M, et al. Contemporary classification of histiocytic disorders. The WHO Committee on Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol. 1997;29:157-66. [ Links ]
7. Gogusev J, Barbey S, Nezelof C. Human malignant histiocytosis CD30+ DEL cell line differentiates into macrophage-like cells when treated with a phorbol diester. Cancer Research. 1991;51(20):5712-5. [ Links ]
8. Kogan SC, Ward JM, Anver MR, Berman JJ, Brayton C, Cardiff RD, et al. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood. 2002;100(1):238-45. [ Links ]
9. Sastre Urgelles A. Hemophagocytic syndromes: Think of them... because they are a reality. An Esp Pediatr. 2002;56:95-8. [ Links ]
10. Torres A, Fernández R, Taylor V, Corona P, Longchong M. Biopsia de médula ósea: Patrones morfológicos de las hemopatías y tumores sólidos en pediatría. Rev Cubana Pediatr. 1996;68(3):171-6. [ Links ]
11. Dearth J, Hunter D, Kelly D, Crist W. Sinus histiocytosis with massive lymphadenopathy. CA Cancer J Clin. 1980;30(1):55-8. [ Links ]
12. Healey PJ, Hight DW. Abdominal masses. En: Wyllie R, Hyams JS, eds. Pediatric gastrointestinal disease. 2a ed. Philadelphia: WB Saunders Company; 1999. p. 281-92. [ Links ]
13. Loredo A. El niño con hepatoesplenomegalia de origen incierto. En: Loredo A, ed. Medicina Interna Pediátrica. 3a ed. México DF: Mc Graw Hill Interamericana; 1997. p. 445-55. [ Links ]
14. Fernandez C, Martinez MA, Ricard P, Ballestin C, Asso A, Hernandez E, et al. Histiocitosis maligna: estudio de 8 casos. Med Clin (Barc). 1988;90:437-42. [ Links ]
15. Reiner AP, Spivak JL. Hematophagic histiocytosis. A report of 23 new patients and a review of the literature. Medicine (Baltimore). 1988;67:369-88. [ Links ]
16. Richard E, Behrman N. Tratado de Pediatria. 14a ed. Madrid: Mc Graw Hill Interamericana; 1992. p. 1225-30, 2141-43. [ Links ]
Dr. Oscar Ruiz Franco
Instituto de Investigaciones Clínicas
Hospital Dos de Mayo
Parque de la Medicina s/n. Lima 1, Perú
Correo-e: osma@ec-red.com
