










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. v.67 n.1 Lima ene./mar. 2006
Hipomagnesemia
Helbert Rondón-Berríos1
1Nephrology Fellow. Renal-Electrolyte Division. Department of Medicine. University of Pittsburgh Medical Center. Pittsburgh, Pennsylvania, USA.
Resumen
El magnesio es el cuarto catión más abundante en el organismo y el segundo catión intracelular más abundante después del potasio. El magnesio es esencial en la transferencia, almacenamiento e utilización de energía, regulando y catalizando más de 300 sistemas enzimáticos. La hipomagnesemia puede por tanto producir una variedad de anormalidades metabólicas y consecuencias clínicas. La hipomagnesemia es un desorden electrolítico común en pacientes hospitalizados. Esta se produce por un desbalance entre la absorción gastrointestinal y la excreción renal de magnesio. La principal manifestación de hipomagnesemia son las arritmias cardíacas que de no ser reconocidas y tratadas pueden ser fatales. La ruta endovenosa de reemplazo de magnesio debe ser usada cuando se requiere corrección urgente de la hipomagnesemia. Los preparados orales deben ser usados para el reemplazo crónico. En este artículo se revisa la hipomagnesemia con énfasis en los mecanismos moleculares responsables de las anormalidades en la homeostasis del magnesio, diagnostico diferencial y tratamiento apropiado.
Palabras clave: Magnesio; deficiencia de magnesio; hipertrofia.
Hypomagnesemia
Abstract
Magnesium is the fourth-most abundant cation in the human body and the second-most abundant intracellular cation after potassium. Magnesium is pivotal in the transfer, storage, and utilization of energy as it regulates and catalyzes more than 300 enzyme systems. Hypomagnesemia may thus result in a variety of metabolic abnormalities and clinical consequences. Hypomagnesemia is a common electrolyte abnormality in hospitalized patients. It results from an imbalance between gastrointestinal absorption and renal excretion of magnesium. The main consequence related directly to hypomagnesemia is cardiovascular arrhythmias and if this not recognized and treated it might be fatal. When urgent correction of hypomagnesemia is required, the intravenous route should be used. Oral magnesium preparations are available for chronic use. In this article I review hypomagnesemia with emphasis on the molecular mechanisms responsible for abnormalities in magnesium homeostasis, differential diagnosis and appropriate therapy.
Keywords: Magnesium; magnesium deficiency; hypertrophy.
INTRODUCCIÓN
El magnesio es el cuarto catión más abundante en el organismo y el segundo catión más abundante en el compartimiento intracelular (1). El magnesio es esencial para la función de muchas enzimas, incluyendo aquellas que se encuentran relacionadas con la transferencia de grupos fosfato, todas las reacciones que requieren ATP y cada paso relacionado con la replicación y trascripción del ADN y la traducción del ARNm. Se estima que el magnesio corporal total constituye unos 1000 mmol o 22,66 g. El 99% del magnesio corporal total está localizado en el compartimiento intracelular. De ese total, el 60% está localizado en el hueso, 20% en el músculo y otro 20% en otros tejidos (1,2). La traslocación de magnesio del compartimiento intracelular al extracelular se realiza de manera lenta, tomando varias semanas. Solamente 1% del magnesio corporal total está ubicado en el compartimiento extracelular. De ese total, 60% se encuentra en forma libre o ionizada, 10% está ligado a sales de citrato, fosfato, oxalato y otros aniones formando complejos y un 30% está ligado a proteínas (2). La concentración de magnesio en el plasma es mantenida en un rango estrecho de 1,7 a 2,2 mg/dL (0,75 a 0,95 mmol/L o 1,5 a 1,9 mEq/L) (1). La homeostasis del magnesio depende del balance entre su absorción intestinal y su excreción renal. La hipomagnesemia es definida como una concentración plasmática de magnesio menor de 1,7 mg/dL (< 0,75 mmol/L o < 1,5 mEq/L. El peso atómico del magnesio es 24,3 (1 mmol/L = 2 mEq/L = 2,4 mg/dL) (1). La hipomagnesemia es un desorden electrolítico común, ocurriendo en alrededor de 12% de pacientes hospitalizados (3). Su incidencia se incrementa hasta 60 a 65% en pacientes críticamente enfermos (4). La presencia de hipomagnesemia en estos pacientes ha sido asociada con un incremento en la morbilidad y mortalidad (5).
FISIOLOGÍA DE LA HOMEOSTASIA DEL MAGNESIO
Absorción de magnesio en el tracto gastrointestinal
En la dieta promedio se ingiere 360 mg (15 mmol) de magnesio elemental. El requerimiento diario de magnesio elemental es 0,15 a 0,20 mmol/kg (1,2). Fuentes ricas en magnesio incluyen los cereales, granos, nueces, legumbres, chocolate, vegetales verdes, algunas carnes y mariscos. Normalmente, solo 50% del magnesio de la dieta es absorbido en el tracto gastrointestinal, primariamente en el yeyuno proximal y el íleo. Alrededor de 40 mg de magnesio al día son también secretados en el intestino y de ellos solo 20 mg son reabsorbidos en el colon y recto (2). La absorción de magnesio en el íleo ocurre a través de 2 procesos. El primero es un proceso activo y saturable, que constituye la ruta principal de transporte de magnesio. Éste se realiza a través del canal de magnesio TRPM6 (transient receptor potential melastatin) 6). Un segundo mecanismo, que es pasivo y no saturable, se realiza a través de la ruta paracelular.
Excreción de magnesio en el riñón (Figura 1)
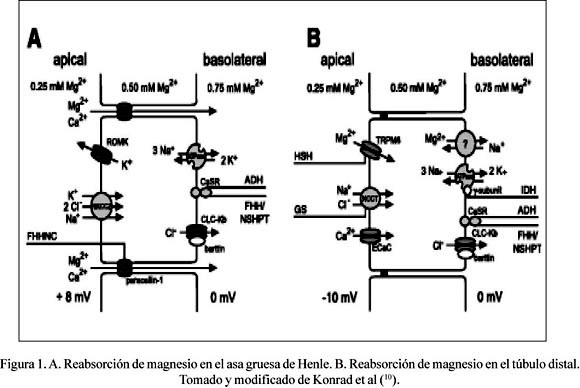
El 80% de magnesio en el plasma es filtrado por el glomérulo, del cual un 95% es reabsorbido por la nefrona. Apro-ximadamente, 100 mg de magnesio son excretados en la orina cada día. A diferencia de otros iones, la absorción tubular de magnesio ocurre principalmente en el asa gruesa de Henle, siendo ésta un 60 a 70% del total filtrado. El túbulo proximal absorbe solo 15 a 25% del magnesio filtrado; por su parte, el túbulo distal absorbe 5 a 10% del magnesio filtrado, pero es considerado como el sitio de control final en la regulación de magnesio (1). En el asa gruesa de Henle, el magnesio es reabsorbido con el calcio de manera pasiva a través de la vía paracelular formada por uniones intercelulares estrechas. La fuerza que impulsa esta reabsorción es la gradiente eléctrica generada por la reabsorción de sodio a través del cotransportador Na+/K+/2Cl- (NKCC2) (6). La paracelina-1, también conocida como claudina-16, ha sido identificada como la proteína constituyente de estas uniones intercelulares estrechas (7). En el túbulo distal, el magnesio es reabsorbido a través de un mecanismo activo, que involucra el canal de magnesio TRPM6 (8). El mecanismo de transporte del magnesio en la membrana baso lateral de las células del asa gruesa de Henle y túbulo distal es desconocido. El transporte de magnesio en esta membrana debiera ser en contra de una gradiente electroquímica. La mayoría de los estudios apuntan hacia un mecanismo de intercambio dependiente de sodio, favorecido por bajas concentraciones de sodio intracelular generadas por la bomba de Na+/K+-ATPasa (9).
Factores que influencian la excreción de magnesio por el riñón
Múltiples son los factores que regulan la excreción de magnesio en el riñón:
Concentración plasmática de magnesio y calcio.
Volumen del extracelular.
Tasa de filtración glomerular.
Estado ácido-base.
Hormonas.
La concentración plasmática de magnesio es el principal regulador de la excreción de magnesio en el riñón. La hipermagnesemia inhibe la reabsorción de magnesio en el asa gruesa de Henle, mientras que la hipo-magnesemia la estimula. La concentración plasmática de calcio tiene un efecto similar. La hipermagnesemia e hipercalcemia inhiben la reabsorción de magnesio a través de la activación del receptor-sensor del calcio (CaSR), un miembro de la familia de receptores ligados a proteína G (9). El CaSR se expresa en la membrana baso lateral de las células del asa gruesa de Henle y túbulo distal. Cuando el magnesio o calcio activan el receptor, se estimula la formación de un derivado del ácido araquidónico, el 20-HETE, el cual de manera reversible inhibe los canales de potasio ROMK1 en el asa gruesa de Henle. La secreción de potasio a través de los canales ROMK1 tiene 2 funciones: primero, provee potasio para la reabsorción de sodio y cloro a través del cotransportador NKCC2; y, segundo, interviene en la producción de la gradiente eléctrica necesaria para la reabsorción pasiva de magnesio y calcio. Por tanto, la inhibición de los canales de potasio ROMK1 en el asa gruesa de Henle reduciría tanto el transporte de sodio como la reabsorción pasiva de magnesio y calcio (11). Otro mecanismo recientemente descubierto es que altas concentraciones de magnesio pueden actuar en la región promotora del gen hPCLN-1, que expresa la proteína paracelina-1, disminuyendo por ende la trascripción de esta proteína y por tanto la reabsorción paracelular de magnesio (12). El volumen del fluido extracelular también influencia la excreción de magnesio. La expansión de volumen inhibe la reabsorción de magnesio en el asa gruesa de Henle, probablemente debido a un incremento en la carga de sodio y por ende una disminución en la gradiente eléctrica, que favorece el transporte paracelular de magnesio.
Cambios en la tasa de filtración glomerular también pueden influenciar la excreción renal de magnesio. Cuando la tasa de filtración glomerular disminuye y, por ende, la carga de magnesio filtrada, la reabsorción de magnesio disminuye.
La depleción de fosfato disminuye la reabsorción de magnesio por un mecanismo desconocido.
La acidosis metabólica crónica produce pérdida renal de magnesio, mientras que la alcalosis metabólica crónica produce el efecto opuesto. La acidosis metabólica crónica disminuye la expresión del canal de magnesio TRPM6 en el túbulo distal, disminuyendo la reabsorción de magnesio a ese nivel. La alcalosis metabólica crónica aumenta la expresión de este canal, produciendo el efecto opuesto (13).
Varias hormonas, incluyendo la 1,25(OH)2 vitamina D, paratohormona, calcitonina, glucagón, aldosterona, hor-mona antidiurética, insulina, prostaglandina E2 y catecolaminas aumentan la reabsorción de magnesio en el asa gruesa de Henle y el túbulo distal. El mecanismo es desconocido, pero se piensa que en muchos casos esto estaría relacionado con el incremento de AMPc a nivel intracelular. Además, estudios recientes demuestran que la 1,25(OH)2 vitamina D3 produciría también un incremento en la expresión de paracelina-1 a través de la activación del factor de transcripción PPAR y su posterior unión al elemento de respuesta específico PPRE en la región promotora del gen hPCLN-1 (12).
FISIOPATOLOGÍA DE LA HIPOMAGNESEMIA
La hipomagnesemia se puede producir por 4 mecanismos fisiopatológicos:
Disminución de la ingesta de magnesio.
Redistribución o translocación de magnesio del extracelular al intracelular.
Pérdida gastrointestinal de magnesio.
Pérdida renal de magnesio.
CAUSAS DE HIPOMAGNESEMIA
Las causas de hipomagnesemia pueden ser agrupadas de acuerdo a su mecanismo fisiopatológico en 4 grupos (Tabla 1).

Disminución de la ingesta
La disminución en la ingesta rara vez causa deficiencia de magnesio, ya que la mayoría de alimentos contiene cantidades significativas de este elemento y el riñón es capaz de adaptarse y conservar magnesio de manera muy eficiente (14). Sin embargo, la hipomagnesemia puede ocurrir en 3 grupos de pacientes: pacientes desnutridos, pacientes alcohólicos y pacientes en los que se administra nutrición parenteral total por tiempos prolongados (15).
Redistribución
La traslocación de magnesio del extracelular al intracelular es una causa frecuente de hipomagnesemia. Esto puede ocurrir en el llamado síndrome del hueso hambriento, en el cual el magnesio se deposita en el hueso. Este síndrome ocurre en pacientes con hiperparatiroidismo después de una paratiroidectomía o en pacientes con hipertiroidismo después de una tiroidectomía.
La hipomagnesemia puede ocurrir también debido a hiperinsulinemia durante el tratamiento de la cetoacidosis diabética, en el síndrome de realimentación o durante la administración intravenosa de dextrosa. En estos casos, la insulina produciría traslocación de magnesio al interior de las células (15,2).
Estados hiperadrenérgicos, como los síndromes de abstinencia al alcohol, pueden incrementar los niveles plasmáticos de catecolaminas, que producirían traslocación de magnesio al intracelular o incrementarían la oxidación de triglicéridos, que a su vez incrementarían los niveles plasmáticos de ácidos grasos libres que quelarían el magnesio plasmático libre (15,2).
La pancreatitis aguda puede causar también hipomagnesemia. El mecanismo probablemente es similar al del asociado a hipocalcemia, esto es la saponificación del magnesio y calcio en la grasa necrótica peripancreática (16).
Pérdida gastrointestinal
Alteraciones de la absorción del magnesio en el intestino pueden ocurrir como consecuencia de diarrea de cualquier causa o resección quirúrgica del intestino. Los pacientes con ileostomías pueden desa-rrollar hipomagnesemia debido a que hay cierta reabsorción de magnesio en el colon.
La hipomagnesemia con hipocalcemia secundaria (HHS) es un desorden autosómico recesivo caracterizado por hipomagnesemia severa asociada con hipocalcemia. La fisiopatología de la hipo-magnesemia en esta entidad está relacionada con un defecto en la reabsorción de magnesio en el intestino y en el túbulo distal. Recientemente, mutaciones en el gen TRPM6, que expresa el canal de magnesio TRPM6, han sido identificadas como el desorden genético subyacente (8).
Pérdida renal
Varios desórdenes tubulares hereditarios son responsables de la pérdida urinaria de magnesio. El síndrome de Gitelman es un trastorno autosómico recesivo causado por mutaciones en el gen SCL12A3 que expresa el cotransportador NaCl (NCCT) en el túbulo distal. Este síndrome se caracteriza por hipopotsemia, hipomagnesemia e hipocalciuria. La hipomagnesemia se encuentra presente en la mayoría de los pacientes con síndrome de Gitelman y en el pasado se asumía que estaba relacionada al defecto en el cotransportador NCCT, pero el mecanismo exacto era desconocido. Recientemente, algunos estudios apuntan a que la pérdida de magnesio se debería a la disminución en la expresión del canal de magnesio TRPM6 en el túbulo distal (17).
De las 5 variantes del síndrome de Bartter, solo el síndrome de Bartter clásico o tipo III está relacionado con hipomagnesemia. Esta variante del síndrome de Bartter es causada por mutaciones en el gen CLCNKB, que expresa el canal de cloro CLC-Kb localizado en la membrana baso lateral del asa gruesa de Henle y túbulo distal. Este canal medía el flujo de cloro al intersticio. Se ignora el mecanismo de la hipomagnesemia en este síndrome (18).
En la hipomagnesemia familiar con hipercalciuria y nefrocalcinosis (HFHNC), un desorden autosómico recesivo, existe una pérdida renal severa de magnesio y calcio. La hipercalciuria a menudo lleva a la nefrocalcinosis, resultando en enfermedad renal crónica. Otros síntomas reportados en pacientes con HFHNC incluyen infecciones del tracto urinario, nefrolitiasis, acidosis tubular renal distal e incompleta y anor-malidades oculares. Este síndrome es causado por mutaciones en el gen hPCLN1 que expresa la proteína paracelina-1 (19).
La hipocalcemia autosómica dominante con hipercalciuria (HADH) constituye otro desorden de pérdida urinaria de magnesio. Los individuos afectados presentan hipocalcemia, hipercalciuria y poliuria, y cerca de 50% de pacientes además presenta hipomagnesemia. LA HADH es producida por mutaciones del gen CaSR, que expresa el CaSR localizado en la membrana baso lateral del asa gruesa de Henle y el túbulo distal. Las mutaciones en este gen alteran el umbral del receptor, incrementando su sensibilidad para el calcio y magnesio. Esto produciría una disminución en la reabsorción de calcio y magnesio en el asa gruesa de Henle y el túbulo distal, produciendo pérdida urinaria de magnesio y calcio (20).
La hipomagnesemia aislada de herencia autosómico dominante (HAHAD) es un desorden asociado con pocos síntomas, excepto por condrocalcinosis. Los pacientes siempre desarrollan hipocalciuria e hipomagnesemia leve. Estudios han identificado una mutación en el gen FXYD2, que expresa la subunidad gamma de la bomba de Na+/K+-ATPasa en la membrana baso lateral del túbulo distal. Esta mutación en la subunidad gama se piensa produce alteraciones en el tráfico de la bomba de Na+/K+-ATPasa hacia la membrana baso lateral, produciendo disminución de la expresión de la misma en la superficie celular. Consecuentemente, la entrada de potasio a la célula se reduce, despolarizando a la célula y cerrando el canal de magnesio TRPM6, produciendo pérdida urinaria de magnesio (21).
En la hipomagnesemia aislada de herencia autosómica recesiva (HAHAR), los individuos afectados presentan síntomas de hipomagnesemia durante la infancia temprana. Se distingue de la forma domi-nante por la ausencia de hipocalciuria. El mecanismo de hipomagnesemia en este síndrome es desconocido (22).
La hipomagnesemia con hipocalcemia secundaria (HHS), también llamada hipomagnesemia intestinal primaria, es un desorden autosómico recesivo que se caracteriza por hipomagnesemia e hipocalcemia severas. Los pacientes se presentan dentro de los 3 primeros meses de vida con síntomas neurológicos de hipomagnesemia e hipocalcemia, inclu-yendo convulsiones, tetania y espasmos musculares. Si no es tratado, el trastorno puede resultar en daño neurológico irreversible o, de otro modo, ser fatal. La hipocalcemia es secundaria al estado de hipoparatiroidismo, debido a la deficiencia de magnesio. Usualmente, la hipocalcemia es resistente a vitamina D. El alivio de los síntomas puede ser alcanzado con la administración de altas dosis de magnesio por vía oral; esto puede llegar a representar hasta 20 veces la ingesta normal. Los estudios han identificado una mutación en el gen TRPM6, que expresa el canal de magnesio TRPM6 (8).
Varios medicamentos, incluyendo diu-réticos de asa como furosemida, bumetanida o ácido etacrínico, producen magnesiuria a través de inhibición del cotransportador NKCC2, que produce la gradiente eléctrica necesaria para la reabsorción de magnesio en el asa gruesa de Henle. El uso crónico de tiazidas puede producir deficiencia de magnesio. El mecanismo se piensa es debido a una disminución en la expresión del canal de magnesio TRPM6 (17).
Muchos medicamentos nefrotóxicos, incluyendo los aminoglicósidos, cisplatino, amfotericina, ciclosporina, tacrolimus y pentamidina, producen magnesiuria por diferentes mecanismos. Por ejemplo, tacrolimus causa hipomagnesemia a través de la disminución en la expresión del canal de magnesio TRPM6 (23). Por otro lado, los aminoglicósidos estimularían la acción del CaSR en el asa gruesa de Henle y túbulo distal, produciendo magnesiuria (24). Algunos estudios sugieren que la hipomagnesemia asociada a cisplatino sería el resultado de la pérdida gastrointestinal de magnesio y no, como se piensa actualmente, a una pérdida renal (25).
Otras causas de hipomagnesemia debido a pérdida renal incluyen hiperaldosteronismo, debido a la expansión de volumen que este produce, hipercalcemia a través de la estimulación del CaSR, hipofosfatemia por razones desconocidas y alcohol, debido a disfunción tubular que es reversible dentro de las 4 semanas de abstinencia.
Finalmente, se puede observar pérdida renal de magnesio en la fase poliúrica de la necrosis tubular aguda o después de una obstrucción (2).
MANIFESTACIONES CLÍNICAS DE HIPOMAGNESEMIA
La mayoría de pacientes con hipomagnesemia no tiene síntomas. Los síntomas de hipomagnesemia no aparecen hasta que la concentración de magnesio plasmática cae por debajo de 1,2 mg/dL. Además, la hipomagnesemia se presenta acompañada por otros desórdenes electrolíticos, como hipopotsemia e hipocalcemia, lo cual hace difícil distinguir las manifestaciones clínicas relacionadas solamente a la deficiencia de magnesio (Tabla 2).

La hipopotsemia es un evento común en pacientes con hipomagnesemia, ocurriendo en 40 a 60% de los casos. En parte, esta se debe a la enfermedad subyacente que causa tanto pérdidas de magnesio como de potasio, lo que sucede por ejemplo en pacientes que toman diuréticos o con diarrea. Pero, en realidad, el principal mecanismo de hipopotsemia debido a hipomagnesemia tiene que ver con las propiedades biofísicas intrínsecas de los canales ROMK1, que median la secreción de potasio en el asa gruesa de Henle. Los canales ROMK1 son canales rectificadores internos de potasio, lo que significa que tienen mayor conductancia para el potasio que fluye hacia dentro de la célula que el que lo hace hacia fuera de la célula. El mecanismo de esta conductancia preferencial hacia dentro de la célula resulta de la unión y bloqueo subsecuente de la conductancia de potasio hacia fuera de la célula por el magnesio intracelular y poliaminas. Una reducción en el magnesio intracelular producto de la deficiencia de magnesio produciría una disminución en la rectificación interna y por ende un incremento de la conductancia de potasio hacia fuera de la célula, con la consiguiente pérdida de potasio e hipopotsemia (26). Cierta evidencia también sugiere que, siendo el magnesio un importante cofactor en la formación de ATP, la hipomagnesemia produciría disminución en la formación de ATP. Se conoce que el ATP es un potente inhibidor de los canales ROMK1. Por tanto, una disminución en el ATP debido a hipomagnesemia produciría pérdida de la inhibición de los canales ROMK1 e incremento de la secreción de potasio (27). De cualquier modo, la hipopotsemia inducida por hipomagnesemia se caracteriza por ser refractaria al tratamiento con suplementos de potasio y solo podrá ser corregida con la corrección de la deficiencia de magnesio.
La hipomagnesemia también puede inducir hipocalcemia. Esto ocurre generalmente cuando la hipomagenesemia es severa (<1,2 mg/dL). El mecanismo es múltiple. Hay una disminución de la liberación de paratohormona. El mecanismo no es conocido al detalle; pero, se piensa que el magnesio incrementaría la actividad de la subunidad alfa de la proteína G relacionada con el CaSR (28). La hipomagnesemia también puede causar resistencia a las acciones de la paratohormona en el tejido óseo. Al parecer la deficiencia de magnesio interfiere con la generación de AMPc, que es el mediador intracelular de la paratohormona.
Enfermedades asociadas a la deficiencia de magnesio
Algunas enfermedades crónicas, como hipertensión arterial, enfermedad coronaria, insuficiencia cardiaca, hipercolesterolemia y diabetes mellitus, han sido asociadas con la deficiencia de magnesio. Una descripción detallada de estas asociaciones está fuera del alcance de esta revisión.
DIAGNÓSTICO DE IPOMAGNESEMIA
Evaluación de la deficiencia de magnesio
La forma más simple de evaluar la deficiencia de magnesio es a través de la medición de la concentración plasmática de magnesio. Con respecto a esto, hay dos consideraciones importantes. Treinta por ciento del magnesio se liga a la albúmina; por tanto, la hipoalbuminemia puede producir una pseudohipomagnesemia. Por otro lado, la mayor cantidad de magnesio en el organismo está localizada en el compartimiento intracelular. Por tanto, una persona puede tener un nivel de magnesio plasmático normal y aún así tener deficiencia de magnesio intracelular y exhibir signos de hipomagnesemia; esto es conocido como deficiencia funcional de magnesio. El magnesio ionizado libre en el plasma es la forma fisiológicamente activa y además la forma que mejor refleja las reservas intracelulares de magnesio. Desafortunadamente, en la práctica clínica actual no se cuenta con un examen de laboratorio que pueda medir niveles plasmáticos de magnesio libre.
Una forma de evaluar la deficiencia funcional de magnesio en pacientes con concentraciones de magnesio plasmático normales, pero en los que se sospecha una deficiencia de magnesio, es por medio de la medición del magnesio plasmático después de una carga de magnesio (29). Primero, se mide la excreción basal de magnesio en orina de 24 h. Luego, se administra una infusión de 7,5 g de sulfato de magnesio en 8 horas y posteriormente se mide la excreción de magnesio en 24 h. Si el paciente excreta < 70% de la carga de magnesio más la excreción de magnesio basal, esto es considerado como deficiencia funcional de magnesio (29).
Diagnosticando la causa de la hipomagnesemia
Si la hipomagnesemia es confirmada, el diagnóstico usualmente se obtiene por anamnesia. Si la causa no es aparente, la distinción entre pérdida renal y pérdida gastrointestinal de magnesio se puede hacer midiendo la cantidad de magnesio en una muestra de orina de 24 horas o calculando la fracción de excreción de magnesio en una muestra de orina obtenida al azar. La fórmula para calcular la fracción de excreción de magnesio (FEMg) es como sigue:
FEMg = UMg x PCr x 100
0,7 x PMg x UCr
Donde UMg y UCr representan las concentraciones urinarias de magnesio y creatinina, respectivamente, y PMg y PCr representan las concentraciones plasmáticas de magnesio y creatinina, respectivamente. La PMg debe ser multiplicada por 0,7, porque solo 70% del magnesio en el plasma está libre y no ligado a proteínas y por tanto es susceptible de ser filtrado por el glomérulo. Una FEMg mayor de 3% o mas de 1 mmol (24 mg) de magnesio en orina de 24 horas indicarían pérdida renal de magnesio (1,2).
TRATAMIENTO DE LA HIPOMAGNESEMIA
En general, los pacientes con hipo-magnesemia deben seguir una dieta rica en magnesio y la causa de hipomagnesemia debe ser tratada, de ser posible.
Si el paciente es asintomático o la hipomagnesemia no es severa (PMg > 1 mg/dL), la vía oral es la ruta de elección, preferiblemente con preparaciones de liberación prolongada, como el cloruro de magnesio o el lactato de magnesio. El óxido de magnesio puede ser usado, pero tiene más efectos adversos gastrointestinales. En los casos leves, no es aconsejable usar la ruta endovenosa, debido a que una elevación abrupta en el magnesio plasmático removería parcialmente el estímulo para la reabsorción de magnesio en el asa gruesa de Henle y hasta un 50% del magnesio infundido sería eliminado en la orina. La repleción de magnesio en estos casos se hace de manera lenta en varios días.
En casos sintomáticos o cuando la concentración de magnesio es < 1 mg/dL, la ruta endovenosa es la preferida. La preparación de elección es el sulfato de magnesio.
Se debe monitorizar los niveles de magnesio plasmático buscando signos de toxicidad, como oliguria, depresión de conciencia y arreflexia. Los pacientes con insuficiencia renal deben recibir el 50% de la dosis si la creatinina sérica es mayor de 2. En casos de toxicidad, el antídoto es cloruro de calcio o gluconato de calcio endovenoso.
Los pacientes con hipomagnesemia inducida por diuréticos que por alguna razón no puedan descontinuarlos pueden beneficiarse del uso de amiloride, el cual puede disminuir la excreción de magnesio en el túbulo distal. Al parecer, amiloride produciría hiperpolarización de la membrana celular, lo cual favorecería la producción del potencial transmembrana necesaria para la reabsorción de magnesio (30).
REFERENCIAS BIBLIOGRÁFICAS
1. Weisinger JR, Bellorin-Font E. Magnesium and phosphorus. Lancet. 1998;352(9125):391-6. [ Links ]
2. Topf JM, Murray PT. Hypomagnesemia and hypermagnesemia. Rev Endocr Metab Disord. 2003;4(2):195-206. [ Links ]
3. Wong ET, Rude RK, Singer FR, Shaw ST Jr. A high prevalence of hypomagnesemia and hypermagnesemia in hospitalized patients. Am J Clin Pathol. 1983;79:348-53. [ Links ]
4. Ryzen E, Wagers PW, Singer FR, Rude RK. Magnesium deficiency in a medical ICU population. Crit Care Med. 1985;13:19-21. [ Links ]
5. Rubeiz GJ, Thill-Baharozian M, Hardie D, Carlson RW. Association of hypomagnesemia and mortality in acutely ill medical patients. Crit Care Med. 1993;21:203-9. [ Links ]
6. Quamme GA. Effect of furosemide on calcium and magnesium transport in the rat nephron. Am J Physiol. 1981;241:F340-F347. [ Links ]
7. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science. 1999;285:103-6. [ Links ]
8. Schlingmann KP, Weber S, Peters M, Niemann Nejsum L, Vitzthum H, Klingel K, et al. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet. 2002;31(2):166-70. [ Links ]
9. Quamme GA. Renal magnesium handling: New insights in understanding old problems. Kidney Int. 1997;52:1180. [ Links ]
10. Konrad M, Schlingmann KP, Gudermann T. Insights into the molecular nature of magnesium homeostasis. Am J Physiol Renal Physiol. 2004;286(4):F599-605. [ Links ]
11. Wang WH, Lu M, Hebert SC. Cytochrome P-450 metabolites mediate extracellular Ca2+-induced inhibition of apical K+ channels in the TAL. Am J Physiol. 1996;271:C103-C111. [ Links ]
12. Efrati E, Arsentiev-Rozenfeld J, Zelikovic I. The human paracellin-1 gene (hPCLN-1): renal epithelial cell-specific expression and regulation. Am J Physiol Renal Physiol. 2005;288:F272-F283. [ Links ]
13. Nijenhuis T, Renkema KY, Hoenderop JG, Bindels RJ. Acid-base status determines the renal expression of Ca2+ and Mg2+ transport proteins. J Am Soc Nephrol. 2006;17(3):617-26. [ Links ]
14. Fitzgerald MG, Fourman P. An experimental study of magnesium deficiency in man. Clin Sci. 1956;15:635-47. [ Links ]
15. Berkelhammer C, Bear RA. A clinical approach to common electrolyte problems: 4. Hypomagnesemia. Can Med Assoc J. 1985;132(4):360-8. [ Links ]
16. Ryzen E, Rude RK. Low intracellular magnesium in patients with acute pancreatitis and hypocalcemia. West J Med. 1990;152(2):145-8. [ Links ]
17. Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest. 2005;115(6):1651-8. [ Links ]
18. Konrad M, Weber S. Recent advances in molecular genetics of hereditary magnesium-losing disorders. J Am Soc Nephrol. 2003;14:249-60 [ Links ]
19. Weber S, Hoffmann K, Jeck N, Saar K, Boeswald M, Kuwertz-Broeking E, et al. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis maps to chromosome 3q27 and is associated with mutations in the PCLN-1 gene. Eur J Hum Genet. 2000;8(6):414-22. [ Links ]
20. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, et al. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med. 1996;335(15):1115-22. [ Links ]
21. Meij IC, Koenderink JB, De Jong JC, De Pont JJ, Monnens LA, Van Den Heuvel LP, et al. Dominant isolated renal magnesium loss is caused by misrouting of the Na+,K+-ATPase gamma-subunit. Ann N Y Acad Sci. 2003;986:437-43. [ Links ]
22. Geven WB, Monnens LA, Willems JL, Buijs W, Hamel CJ. Isolated autosomal recessive renal magnesium loss in two sisters. Clin Genet. 1987;32:398-402. [ Links ]
23. Nijenhuis T, Hoenderop JG, Bindels RJ. Downregulation of Ca(2+) and Mg(2+) transport proteins in the kidney explains tacrolimus (FK506)-induced hypercalciuria and hypomagnesemia. J Am Soc Nephrol. 2004;15(3):549-57. [ Links ]
24. Chou CL, Chen YH, Chau T, Lin SH. Acquired Bartter-like syndrome associated with gentamicin administration. Am J Med Sci. 2005;329(3):144-9. [ Links ]
25. Lajer H, Kristensen M, Hansen HH, Christensen S, Jonassen T, Daugaard G. Magnesium and potassium homeostasis during cisplatin treatment. Cancer Chemother Pharmacol. 2005;55(3):231-6. [ Links ]
26. Nichols CG, Ho K, Hebert SC. Mg2+-dependent inward rectification of ROMK1 potassium channels expressed in Xenopus oocytes. J Physiol. 1994;476:399-409. [ Links ]
27. Hebert SC, Desir G, Giebisch G, Wang W. Molecular diversity and regulation of renal potassium channels. Physiol Rev. 2005;85:319-71. [ Links ]
28. Quitterer U, Hoffmann M, Freichel M, Lohse MJ. Paradoxical block of parathormone secretion is mediated by increased activity of G alpha subunits. J Biol Chem. 2001;276(9):6763-9. [ Links ]
29. Herbert P, Mehta N, Wang J, Hindmarsh T, Jones G, Cardinal P. Functional magnesium deficiency in critically ill patients identified using a magnesium-loading test. Crit Care Med. 1997;25:749–55. [ Links ]
30. Dai LJ, Ritchie G, Kerstan D, Kang HS, Cole DE, Quamme GA. Magnesium transport in the renal distal convoluted tubule. Physiol Rev. 2001;81(1):51-84. [ Links ]
Manuscrito recibido el 02 de marzo de 2006 y aceptado para publicación el 28 marzo de 2006.
Correspondencia: Dr. Helbert Rondon Berrios, MD
Renal-Electrolyte Division
University of Pittsburgh Medical Center
A915 Scaife Hall
3550 Terrace St
Pittsburgh, PA 15261
USA
Correo-e: rondonberriosh@upmc.edu

