










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. v.69 n.3 Lima sep. 2008
Displasia mesenquimal placentaria
Placental mesenchymal dysplasia
Erasmo Huertas1,2, Elías Alexis Valladares3, Guiselle Gutiérrez2,4, Vilma Oros2
1 Unidad de Medicina Fetal, Instituto Nacional Materno Perinatal. Lima, Perú.
2 Facultad de Medicina, Universidad Nacional Mayor de San Marcos. Lima, Perú.
3 Servicio de Emergencia, Instituto Nacional Materno Perinatal. Lima, Perú.
4 Servicio de Patología, Instituto Nacional Materno Perinatal. Lima, Perú.
Resumen
La displasia mesenquimal placentaria es una rara condición de placentomegalia y vellosidades coriales anormales, con frecuencia confundida clínicamente como mola hidatidiforme parcial. Sin embargo, es clínica y patológicamente distinta, con alta incidencia de restricción de crecimiento intrauterino y muerte fetal. Se presenta la evolución clínica de una gestante portadora de una displasia mesenquimal placentaria, el primer caso diagnosticado en el Instituto Nacional Materno Perinatal. Se describe el problema, para sospechar el diagnóstico y el manejo de la gestación. Hubo necesidad de realizar cesárea a las 27 semanas de gestación, obteniéndose una recién nacida mujer de 1 048 gramos, con Ápgar 8 al minuto y 9 a los 5 minutos, líquido amniótico sanguinolento en regular cantidad y placentomegalia. La paciente evolucionó satisfactoriamente y salió de alta al tercer día. En el neonato se confirmó una tumoración abdominal hepática y tuvo que ser intervenido quirúrgicamente, previa ganancia ponderal, determinándose el diagnóstico anatomopatológico de hamartoma mesenquimal de hígado.
Palabras clave: Placenta; ultrasonografía; recién nacido; hamartoma; hígado; patología.
Abstract
Placental mesenchymal dysplasia is a rare condition of placentomegaly and abnormal chorionic villous, often clinically confused as partial hydatidiform mole. However, it is different clinically and pathologically, with high incidence of intrauterine growth restriction and stillbirth. We present the clinical course of a pregnant woman carrying placental mesenchymal dysplasia, the first case diagnosed at the Maternal Perinatal National Institute, how to suspect diagnosis and gestational care. There was need for caesarean section at 27 weeks gestation, obtaining a 1 048 g newborn girl, with Apgar 8 at the first minute and 9 at the fifth minute, bloody amniotic fluid in regular quantity and placentomegaly. Patient evolved satisfactorily and was discharged on the third hospitalization day. An abdominal liver tumor was confirmed in the newborn and surgical pathology report was liver mesenchymal hamartoma.
Key words: Placenta; ultrasonography; infant, newborn; hamartoma; liver; pathology.
INTRODUCCIÓN
La displasia mesenquimal placentaria es una patología placentaria, que por su apariencia macroscópica y ecográfica puede ser confundida con mola hidatidiforme parcial. Se caracteriza por edema de la vellosidad troncal, que origina placentomegalia, pero con desarrollo normal del trofoblasto, sin relación con enfermedad trofoblástica gestacional (1). Esta anomalía es una malformación limitada al mesénquima extraembrionario que envuelve al mesodermo y los vasos de la vellosidad troncal de varios cotiledones (2). Se le ha encontrado asociada al síndrome de Beckwith-Wiedemann (3), hamartoma mesenquimal hepático fetal (4), restricción del crecimiento intrauterino (RCIU) (5), muerte fetal intrauterina (6), placenta previa (7), feto anatómicamente normal asociado con cariotipo 46XX (8), hiperplasia adrenal congénita fetal (9) y malformación de la pared abdominal anterior fetal (10). Una de las principales características de esta anomalía es la presencia de vasos vellosos troncales aneurismales, los cuales, en cortes histológicos de 4-5 µm, pueden ser confundidos con otras alteraciones vasculares, como corioangioma, corioangiosis o corioangiomatosis, originadas en la misma región de la placa coriónica. La dificultad para el diagnóstico histopatológico se incrementa cuando estas últimas alteraciones se presentan simultáneamente con la hiperplasia mesenquimal, como ocurre en casos muy raros (11), en los cuales la microscopia electrónica de barrido aclara el panorama.
Se desconoce la incidencia de esta anomalía vascular; sin embargo, un estudio aislado realizado en Japón, indica una cifra de 0,02% (12). En vista de la importancia de los conocer aspectos clínicos e histopatológicos de esta entidad, describimos el primer caso de displasia mesenquimal placentaria registrado en el Instituto Nacional Materno Perinatal de Lima-Perú.
PRESENTACIÓN DEL CASO
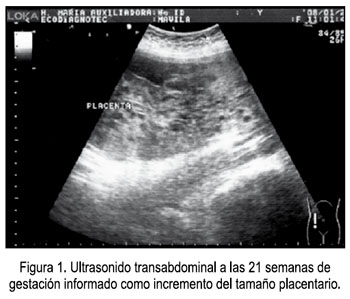
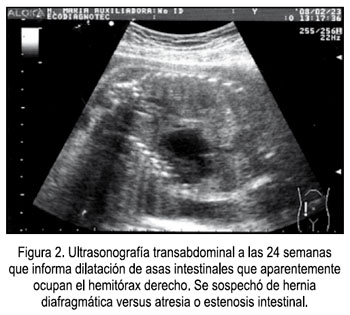
Paciente primigesta de 19 años, con 27 semanas de edad gestacional, sin antecedentes de importancia, controlada en un Hospital de Nivel II, donde le realizaron 3 evaluaciones ecográficas. En la primera, del 18 de diciembre de 2007, se diagnosticó una gestación de 15 semanas sin alteraciones. La segunda ecografía, a las 21 semanas, el 26 de enero de 2008, encontró como único dato relevante placenta aumentada de tamaño, corporal posterior, grado 0 (Figura 1). En el control realizado aproximadamente un mes después (23/02/08), se describe una aparente dilatación de asas intestinales, que ocupan el hemitórax derecho, sospechándose de hernia diafragmática versus atresia o estenosis intestinal (Figura 2). La placenta fue descrita como corporal posterior, sin anomalías aparentes. A los cinco días se repitió el examen, pero esta vez se informó la presencia de una formación quística unilocular en tórax, de 45 x 41 mm, que desplazaba el corazón y no impresionaba ser contenido abdominal, placenta grande grado I, que a nivel de fondo semejaba mola, por lo que se concluyó con el diagnóstico de mola parcial.
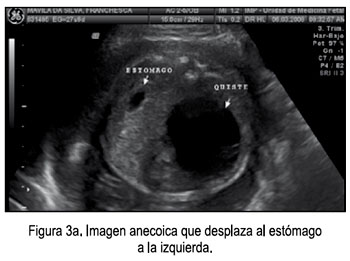
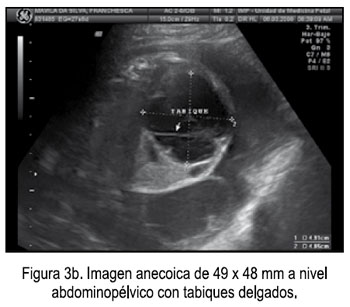
El 6 de marzo acudió al Instituto Nacional Materno Perinatal de la ciudad de Lima, para una evaluación por la Unidad de Medicina Fetal, observándose imagen anecoica, de 49 x 48 mm, a nivel abdominopélvico, con tabiques delgados, que desplaza al estómago a la izquierda, feto de sexo femenino y placenta con degeneración hidrópica (Figuras 3a - 3b, 4), por lo que se planteó los diagnósticos: 1) Gestación única activa de 26,3 semanas, 2) Descartar displasia mesenquimal placentaria versus mola parcial; 3) Quiste abdomino-pélvico de etiología a determinar, D/C ovárico, y se solicita cariotipo fetal y punción de quiste.
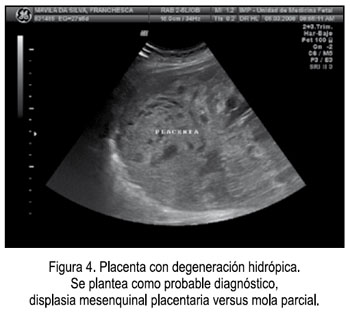
El 7 de marzo se realizó la amniocentesis y se obtuvo 20 mL de líquido amniótico sanguinolento, que se envió para cultivo de células amnióticas. Dos días después (el 9 de marzo), a horas 02:20 am, la paciente ingresó por el Servicio de Emergencia, refiriendo contracciones uterinas y pérdida de líquido amniótico de aproximadamente 20 horas, siendo hospitalizada para tocólisis por los diagnósticos de: 1) Primigesta de 27 semanas por fecha de última menstruación, 2) Trabajo de parto pretérmino, 3) Feto con malformación abdominal, y 4) Placenta con degeneración hidrópica versus mola parcial. Debido al fracaso de la terapia tocolítica, se decidió terminar el embarazo por cesárea, siendo los hallazgos recién nacida mujer, peso 1 048 gramos, Ápgar 81 - 95, líquido amniótico sanguinolento en regular cantidad y placentomegalia.
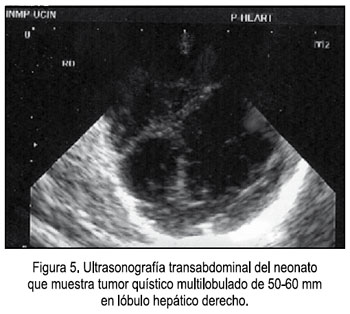
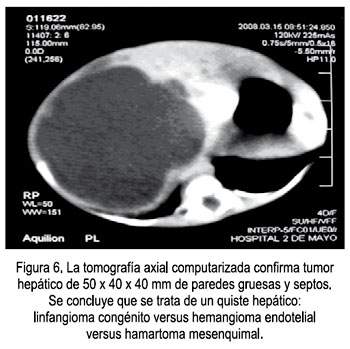
En la recién nacida se constató presencia de tumor blando depresible, al parecer quístico, de 70 x 50 mm de diámetro, móvil, que abarcaba hipocondrio, flanco derecho, epigastrio y mesogastrio, solicitándose ecografía y cariotipo de sangre neonatal. La ecografía mostró un tumor quístico multilobulado, de 50 a 60 mm, en lóbulo hepático derecho (Figura 5) y en la tomografía se observó un tumor hepático de 50 x 40 x 40 mm, de paredes gruesas y septos (Figura 6). Se concluyó que se trataba de un quiste hepático: linfangioma congénito versus hemangioma endotelial versus hamartoma mesenquimal. El neonato fue intervenido quirúrgicamente, previa ganancia ponderal, y el examen anatomo patológico determinó al examen macroscópico: Múltiples fragmentos de tejido, con márgenes de aspecto quístico, con medidas 4,5x3x2 cm; 4x2,5x1,5 cm y 13x3x1,5 cm. Al corte de los quistes se observó paredes traslúcidas, de contenido mucinoso. Microscópicamente, se hizo el diagnóstico de hamartoma mesenquimal de hígado: tumoraciones quísticas que contenían un material gelatinoso formado a partir de células mixoides, el cual rodea grupos de hepatocitos en la periferie y los diseca; no se observó cubierta epitelial, sino tejido fibroso laxo que rodeaba al material mixoide y que se continuaba con el tejido hepático. Lamentablemente, el neonato no toleró el acto quirúrgico, falleciendo en el postoperatorio inmediato. Los padres no autorizaron la realización de la necropsia.
De la muestra de líquido amniótico, el cultivo de células amnióticas no creció. La muestra de sangre neonatal para cariotipo se coaguló, por lo que no fue posible determinar el cariotipo fetal.
El estudio histopatológico de la placenta reveló:
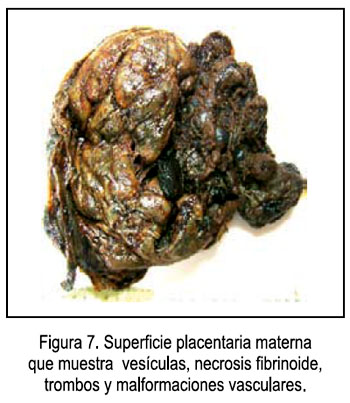
- Características macroscópicas (Figura 7): Peso: 973 gramos, 19 x 17 x 5 centímetros, membranas completas, opacas; cordón de 11 x 0,5 cm, excéntrico, violáceo, espiralado; cara fetal hemorrágica, circunmarginal 30%, trombos en vasos coriales; cara materna rojo vinoso, cotiledones incompletos, sin coágulos, con vesículas de hasta 2 cm de diámetro y depósitos fibrinoides subcoriales.
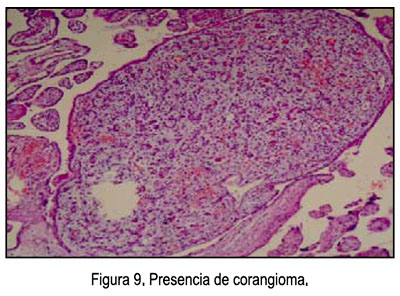
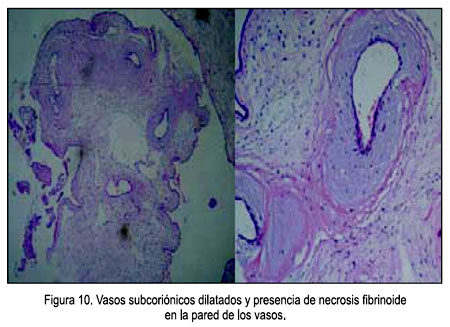
- Características microscópicas (Figuras 8, 9 y 10): Placenta del tercer trimestre, grande para el peso fetal, vellosidades coriales con proliferación de mesénquima, vasos sanguíneos, corioangiosis y edema; cordón con edema; membranas sin alteraciones.
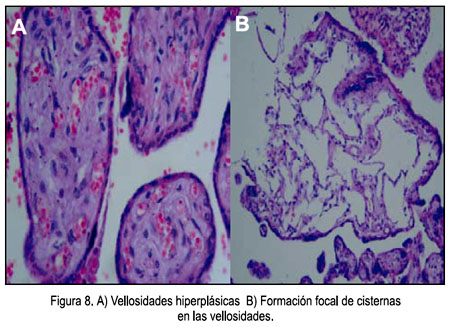
La displasia mesenquimal placentaria (DMP) es una anomalía vascular placentaria rara, de reconocimiento relativamente reciente, caracterizada por placentomegalia y vesículas en racimos que semejan un embarazo molar parcial, al ultrasonido y al examen macroscópico de la placenta.
La displasia mesenquimal placentaria fue descrita inicialmente por Moscoso y col. (1) como hiperplasia vellosa, en 1991, cuando identificó dos casos con niveles elevados de alfafetoproteína materna sérica y placentas grandes, que se correspondían con características ultrasonográficas de mola parcial. Las placentas muestran dilatación aneurismal de los vasos en la cara fetal y vellosidades dilatadas, con material gelatinoso en la región subcoriónica. Sin embargo, histológicamente, estas placentas pueden distinguirse de las molas parciales a causa de la ausencia de proliferación trofoblástica (1). Desde entonces, esta entidad está siendo descrita en forma creciente.
Varios autores han preferido usar el término hiperplasia mesenquimal placentaria, en lugar de displasia (5,11,13,14). La verdadera incidencia de DMP se desconoce, porque previamente ha sido informada bajo una variedad de nombres, como placentomegalia con hydrops masivo de las vellosidades placentarias y molas pseudoparciales (15,16). Además, la DMP, por ser de conocimiento reciente aún, es subdiagnosticada y subregistrada por los patólogos.
Hasta la fecha, se ha publicado en la literatura 82 casos de displasia mesenquimal placentaria, con una preponderancia femenina definitiva de 3,6:1 (mujer:varón) (17), lo que se confirma también en este caso. Al respecto, Yamada y col. (18), señalaron que el gen D del factor de crecimiento endotelial (VEGF-D), o factor relacionado a la angiogénesis, está localizado en el cromosoma Xp22.31 e informaron un caso de DMP en un feto femenino con deleción del brazo corto del cromosoma X.
Paradinas y col. (16) estimaron la frecuencia de una DMP en cada 500 casos, referidos como probables embarazos molares. Según Arizawa y Nakayama (12), la incidencia de DMP es 0,02%.
La displasia mesenquimal placentaria en la mayoría de casos está asociada con el síndrome de Beckwith-Wiedemann y con restricción del crecimiento intrauterino (RCIU), pero también con fetos de apariencia normal. Esta entidad debería ser incluida en el diagnóstico diferencial de lesiones placentarias quísticas por sonografía, especialmente cuando se identifica un feto de apariencia fenotípicamente normal.
Se observa una elevada frecuencia de RCIU. De los fetos sin el síndrome de Beckwith-Wiedemann, el 50% tiene RCIU y muchos pesan menos del percentil 5. De igual manera, se asocia con una tasa alta de muerte fetal y neonatal; aproximadamente, 40% de los fetos y neonatos afectados muere prematuramente, muchos de ellos sin RCIU, lo que implica la contribución de otros factores (19-22).
La placenta tiene un rol importante en la salud fetal, como fuente de nutrientes para el feto, y es la primera línea de defensa. Los casos de displasia mesenquimal placentaria, restricción de crecimiento intrauterino y muerte fetal pueden explicarse por una hipoxia crónica potencialmente secundaria a trombosis vascular obstructiva fetal y disminución del intercambio gaseoso materno-fetal, debido a una cantidad anormal de vellosidades coriónicas normales y cortocircuitos sanguíneos en la superficie de intercambio. Se desconoce las secuelas en infantes con DMP y no se dispone del seguimiento clínico de la mayoría de casos.
Una característica importante de la displasia mesenquimal placentaria consiste en los vasos trombosados (venas y arterias) (23). La dilatación marcada de estos vasos causaría distorsión significativa de los canales vasculares y alteración del flujo dinámico, conduciendo a lesión endotelial y trombosis. Es posible que ciertas anomalías del cordón umbilical, que incluyen inserción marginal e hipertorsión, contribuyan a las anomalías vasculares de la DMP, pudiendo causar obstrucción completa del retorno venoso. Hipotéticamente, si los problemas del cordón no son letales y persisten, la hipoxia fetal resultante estimularía la remodelación de la placenta, como hipervascularidad (corioangiosis y corioangiomas), aumentaría la eritropoyesis y la hiperplasia estromal (vellosidades displásicas).
La patogénesis de la DMP no es clara. La mayoría de casos tiene ultrasonografías anormales, con impresión de molas hidatidiformes, en el segundo o tercer trimestre. Aunque la DMP imita al embarazo molar parcial sonográficamente y al examen macroscópico, una diferencia clave entre las placentas molares y normales es su composición genética. Las molas parciales son 70 a 80% triploides, frecuentemente con dos series de genes paternos y una de genes maternos, como resultado de fertilización dispérmica (24,25). En la DMP, se encuentra con frecuencia cariotipos aparentemente normales.
La constitución genética de estos casos es controversial. Las comunicaciones previas indicaron constitución genética biparental normal (11,14). Sin embargo, recientemente, Cohen y col. (26) describieron 3 casos con aneuplodías: una trisomía 13 con translocación balanceada entre los cromosomas 1 y 13, un síndrome de Klinefelter (47,XXY) y una triplodia (69,XXX; un caso previamente informado por Paradinas y col. (16)). Es posible que estas raras aneuploidias sean coincidencias. La constitución genética de la DMP aún no ha sido establecida, como Kaiser-Rogers y col. (27) los señalan en un informe reciente. Ellos describen dos casos en los que identificaron mosaicismo androgénico/biparental en las placentas con DMP. Estos autores postularon que tales mosaicismos surgen como resultado de la falla del genoma materno para duplicarse antes del primer clivaje, con duplicación normal y segregación del genoma paterno, lo que resulta en dos células hijas, una con genes biparentales normales y una solo con genes paternos (23 cromosomas). La última célula hija se vuelve diploide por endorreduplicación. La célula biparental presumiblemente da origen al feto, amnios y trofoblasto y, la célula androgénica, a una porción variable del corion y mesénquima velloso, con características morfológicas alteradas de DMP. Esta nueva teoría también cuenta para la marcada predominancia femenina en DMP, porque una línea celular 46,YY androgénica presumiblemente no es viable. En este sentido, la DMP estaría relacionada con una mola completa, que con frecuencia es 46,XX androgénica. Este nuevo hallazgo del mosaicismo biparental/androgénico en DMP requiere ser estudiado. Aunque la mayoría de casos de DMP se asocian con fetos completamente normales, algunos casos de DMP con hemangiomas congénitos, hamartomas vascuales y hamartomas hepáticos mesenquimales sugieren que existe un defecto común en el mesodermo placentario y embrionario (4,28).
No se ha descrito una mutación genética para la DMP. Sin embargo, el hallazgo de DMP en fetos con síndrome de Beckwith-Wiedemann sugiere que podría existir una relación genética, porque el síndrome es una condición de sobrecrecimiento constitucional, con asociación genética al cromosoma 11p15.5 (29).
Como se precisó en la discusión, es importante realizar un estudio completo de la placenta, el feto y ambos padres, para avanzar en la investigación de esta entidad, requiriéndose el trabajo multidisciplinario en equipo, que incluye al patólogo, neonatólogo y ginecoobstetra.
REFERENCIAS BIBLIOGRÁFICAS
1. Moscoso G, Jauniaux E, Hustin J. Placental vascular anomaly with diffuse mesenchymal stem villous hyperplasia. A new clinico-pathological entity? Pathol Res Pract. 1991;187:324-8. [ Links ]
2. Jauniaux E. Partial moles: From postnatal to prenatal diagnosis. Placenta. 1999;20:379-88. [ Links ]
3. McCowan LME, Bencroft DM. Beckwith-Wiedemann syndrome, placental abnormalities, and gestational proteinuric hypertension. Obstet Gynecol. 1994;83:813-7. [ Links ]
4. Kitano Y, Ruchelli E, Weiner S, Adzick NS. Hepatic mesenchymal hamartoma associated with mesenchymal stem villous hyperplasia of the placenta. Fetal Diagn Ther. 2000;15:134-8. [ Links ]
5. KuwabaraY, ShimaY, Araki T, Shin S. Mesenchymal stem villous hyperplasia of the placenta and fetal growth restriction. Obstet Gynecol. 2001;98:940-3. [ Links ]
6. Ohyama M, Kojyo T, Gotoda H, Sato T, Ijiri R, Tanaka Y. Mesenchymal dysplasia of the placenta. Pathol Int. 2000;50:759-64. [ Links ]
7. Matsui H, Litsuka Y, Yamazawa K, Tanaka N, Mitsuhashi A, Seki K, et al. Placental mesenchymal dysplasia initially diagnosed as partial mole. Pathol Int. 2003;53:810-3. [ Links ]
8. Jauniaux E, Nicolaides KH, Hustin J. Perinatal features associated with placental mesenchymal dysplasia. Placenta. 1997;18:701-6. [ Links ]
9. Furuhashi M, Oda H, Nakashima T. Hydrops of placental stem villi complicated with fetal congenital adrenal hyperplasia. Arch Gynecol Obstet. 2000;264:101-4. [ Links ]
10. Pridmore BR, Kong TY, Wells WA. Ultrasound placental cysts associated with massive placental stem villous hydrops, diploid DNA content, and exomphalos. Am J Perinatol. 1994;11:14-8. [ Links ]
11. Chen CP, Chern SR, Wang TY, Huang ZD, Huang MC, Chuang CY. Pregnancy with concomitant chorangioma and placental vascular malformation with mesenchymal hyperplasia. Human Reprod. 1997;12:2553-6. [ Links ]
12. Arizawa M, Nakayama M. Suspected involvement of the X chromosome in placental mesenchymal dysplasia. Congenit Anom. 2002;42:309-17. [ Links ]
13. Laberge JM, Patenaude Y, Desilets V, Cartier L, Khalife S, Jutras L, et al. Large hepatic mesenchymal hamartoma leading to mid-trimester fetal demise. Fetal Diagn Ther. 2005;20:141-5. [ Links ]
14. Hojberg KE, Aagaard J, Henriques U, Sunde L. Placental vascular malformation with mesenchymal hyperplasia and a localized chorioangioma: a rarity simulating partial mole. Pathol Res Pract. 1994;190:808-13. [ Links ]
15. Lage JM. Placentomegaly with massive hydrops of placental stem villi, diploid DNA content and fetal omphaloceles: possible association with Beckwith-Wiedemann syndromes. Hum Pathol. 1991;22:591-7. [ Links ]
16. Paradinas FJ, Sebire NJ, Fisher RA, Rees HC, Foskett M, Seckl MJ, et al. Pseudo-partial moles: placental stem vessel hydrops and the association with Beckwith-Wiedemann syndrome and complete moles. Histopathology. 2001;39:447-54. [ Links ]
17. Pham T, Seele J, Stayboldt C, Chan L, Benirschke K. Placental Mesenchymal Dysplasia Is Associated With High Rates of Intrauterine Growth Restriction and Fetal Demise A Report of 11 New Cases and a Review of the Literature. Am J Clin Pathol. 2006;126:67-78. [ Links ]
18. Yamada Y, Nezu J, Shimane M, Hirata Y. Molecular cloning of a novel vascular endothelial growth factor, VEGF-D. Genomics. 1997;42:483-8. [ Links ]
19. Illanes S, Soothill P. Management of fetal growth restriction. Semin Fetal Neonatal Med. 2004;9:395-401. [ Links ]
20. Monk D, Moore G. Intrauterine growth restriction: genetic causes and consequences. Semin Fetal Neonatal Med. 2004;9:371-8. [ Links ]
21. Brodsky D, Christou H. Current concepts in intrauterine growth restriction. J Intensive Care Med. 2004;19:307-19. [ Links ]
22. Parveen Z, Tongson-Ignacio J, Fraser K, Killeen, Thompson K. Placental Mesenchymal Dysplasia. Arch Pathol Lab Med. 2007;131:131-7. [ Links ]
23. Sander C. Angiomatous malformation of placental chorionic stem vessels and pseudo-partial molar placentas: report of five cases. Pediatr Pathol. 1993;13:621-33. [ Links ]
24. Ohama K, Ueda K, Okamoto E, Takenaka M, Fujiwara A. Cytogenetic and clinicopathologic studies of partial moles. Obstet Gynecol. 1986;68:259-62. [ Links ]
25. Jauniaux E, Kadri R, Hustin J. Partial mole and triploidy: screening patients with first-trimester spontaneous abortion. Obstet Gynecol. 1996;88:616-9. [ Links ]
26. Cohen MC, Roper EC, Sebire NJ, Stanek J, Anumba DO. Placental mesenchymal dysplasia associated with fetal aneuploidy. Prenat Diagn. 2005;25:187-92. [ Links ]
27. Kaiser-Rogers KA, McFadden DE, Livasy CA, Dansereau J, Jiang R, Knops JF, et al. Androgenetic/biparental mosaicism causes placental mesenchymal dysplasia. J Med Genet. 2006;43:187-92. [ Links ]
28. Gibson BR, Muir-Padilla J, Champeaux A, Suarez ES. Mesenchymal dysplasia of the placenta. Placenta. 2004;25:671-2. [ Links ]
29. Ping AJ, Reeve AE, Law DJ, Young MR, Boehnke M, Feinberg AP. Genetic linkage of Beckwith-Wiedemann syndrome to 11p15. Am J Hum Genet. 1989;44(5):720-3. [ Links ]
Manuscrito recibido el 1 de setiembre de 2008 y aceptado para publicación el 30 de setiembre de 2008.
Correspondencia:
Dr. Erasmo Huertas Tacchino
Calle Carlos Pane 143
Lima 5, Perú
Correo-e: erasmohuertas@hotmail.com

